ABOUT AUTHORS:
Salma S. Quadri*, Lalit V. Sonwane, Bhagwat N. Poul, Sharada N. Kamshette
Department of Quality Assurance,
MSS’s Maharashtra College of Pharmacy, Nilanga,
Latur, Maharashtra, India.
[email protected]
ABSTRACT
The main contemporary goal of stability indicating methods is to provide information about condition for stress testing so as to establish the stability of drug substances and product. This paper reviews the regulatory aspects for development of stability indicating methods. SIMs are used to differentiate the API from its potential decomposition product. Regulatory guidance in ICH Q1A (R2) ICH Q3B (R2) Q6A and FDA 21 CFR section 211 requires validated stability indicating methods. Force degradation is required to demonstrate the specificity when developing SIMs and for this reason, it should be perform prior to implementing the stability studies. Force degradation of drug standard and excipients is carried out under different conditions to determine whether the analytical method is stability indicating. The approaches for the development of stability indicating method is discussed.
INTRODUCTION
Chemical stability of pharmaceutical molecules is a matter of great concern as it affects the safety and efficacy of the drug product. The FDA and ICH guidance states the requirement of stability testing data to understand how the quality of a drug substance and drug product changes with time under the influence of various environmental factors. Knowledge of the stability of molecule helps in selecting proper formulation and package as well as providing proper storage conditions and shelf life, which is essential for regulatory documentation. Forced degradation is a process that involves degradation of drug products and drug substances at conditions more severe than accelerated conditions and thus generates degradation products that can be studied to determine the stability of the molecule. The ICH guideline states that stress testing is intended to identify the likely degradation products which further helps in determination of the intrinsic stability of the molecule, establishing degradation pathways and to validate the stability indicating procedures used [1]. But these guidelines are very general in conduct of forced degradation and do not provide details about the practical approach towards stress testing. Although forced degradation studies are a regulatory requirement and scientific necessity during drug development, it is not considered as a requirement for formal stability program. It has become mandatory to perform stability studies of new drug moiety before filing in registration dossier. The stability studies include long term studies (12 months) and accelerated stability studies (6 months). But intermediate studies (6 months) can be performed at conditions milder than that used in accelerated studies. So the study of degradation products like separation, identification and quantitation would take even more time. As compared to stability studies, forced degradation studies help in generating degradants in much shorter span of time, mostly a few weeks. The samples generated from forced degradation can be used to develop stability indicating method which can be applied latter for the analysis of samples generated from accelerated and long term stability studies. This review provides a proposal on the practical performance of forced degradation and its application for the development of stability indicating method. The stability-indicating assay is a method that is employed for the analysis of stability samples in pharmaceutical industry. With the advent of International Conference on Harmonisation (ICH) guidelines, the requirement of establishment of stability-indicating assay method (SIAM) has become more clearly mandated. The guidelines explicitly require conduct of forced decomposition studies under a variety of conditions, like pH, light, oxidation, dry heat, etc. and separation of drug from degradation products. The method is expected to allow analysis of individual degradation products. A review of literature reveals a large number of methods reported over the period of last 3–4 decades under the nomenclature ‘stability-indicating’. However, most of the reported methods fall short in meeting the current regulatory requirements. Accordingly, the purpose of this write-up is to suggest a systematic approach for the development of validated SIAMs that should meet the current ICH and regulatory requirements. The discussion also touches upon various critical issues, such as the extent of separation of degradation products, establishment of mass balance, etc., which are important with respect to the development of stability-indicating assays, but are not yet fully resolved. Some other aspects like suitability of pharmacopoeial methods for the purpose and the role of SIAMs in stability evaluation of biological/ biotechnological substances and products are also delved upon.
According to FDA guideline (Guidance for Industry, Analytical Procedures and Methods Validation, FDA, 2000), a Stability Indicating Method (SIM) is defined as a validated analytical procedure that accurate and precisely measure active ingredients (drug substance or drug product) free from process impurities, excipients and degradation products. The FDA recommends that all assay procedures for stability should be stability indicating. The main objective of a stability indicating method is to monitor results during stability studies in order to guarantee safety, efficacy and quality. It represents also a powerful tool when investigating out-of-trend (OOT)[2] or out-of-specification (OOS) [3] results in quality control processes.
REGULATORY STATUS OF STABILITY-INDICATING ASSAYS
The ICH guidelines have been incorporated as law in the EU, Japan and in the US, but in reality, besides these other countries are also using them. As these guidelines reflect the current inspectional tendencies, they carry the de facto force of regulation. The ICH guideline Q1A on Stability Testing of New Drug Substances and Products [4] emphasizes that the testing of those features which are susceptible to change during storage and are likely to influence quality, safety and/or efficacy must be done by validated stability-indicating testing methods. It is also mentioned that forced decomposition studies (stress testing) at temperatures in 10 °C increments above the accelerated temperatures, extremes of pH and under oxidative and photolytic conditions should be carried out on the drug substance so as to establish the inherent stability characteristics and degradation pathways to support the suitability of the proposed analytical procedures. The ICH guideline Q3B entitled ‘Impurities in New Drug Products’ emphasizes on providing documented evidence that analytical procedures are validated and suitable for the detection and Quantitation of degradation products [5]. It is also required that analytical methods should be validated to demonstrate that impurities unique to the new drug substance do not interfere with or are separated from specified and unspecified degradation products in the drug product. The ICH guideline Q6A, which provides note for guidance on specification [6] also, mentions the requirement of stability-indicating assays under Universal Tests/Criteria for both drug substances and drug products. The same is also a requirement in the guideline Q5C on Stability Testing of Biotechnological/Biological Products [7]. Since there is no single assay or parameter that profiles the stability characteristics of such products, the onus has been put on the manufacturer to propose a stability-indicating profile that provides assurance on detection of changes in identity, purity and potency of the product. Unfortunately, none of the ICH guidelines provides an exact definition of a stability-indicating method. Elaborate definitions of stability-indicating methodology are, however, provided in the United States-Food and Drug Administration (US-FDA) stability guideline of 1987 [8] and the draft guideline of 1998 [9]. Stability-indicating methods according to 1987 guideline were defined as the ‘quantitative analytical methods that are based on the characteristic structural, chemical or biological properties of each active ingredient of a drug product and that will distinguish each active ingredient from its degradation products so that the active ingredient content can be accurately measured.’This definition in the draft guideline of 1998 reads as: ‘validated quantitative analytical methods that can detect the changes with time in the chemical, physical, or microbiological properties of the drug substance and drug product, and that are specific so that the contents of active ingredient, degradation products, and other components of interest can be accurately measured without interference.’ The major changes brought in the new guideline are with respect to (i) introduction of the requirement of validation, and (ii) the requirement of analysis of degradation products and other components, apart from the active ingredients(s). The requirement is also listed in World Health Organization (WHO), European Committee for Proprietary Medicinal Products and Canadian Therapeutic Products Directorate’s guidelines on stability testing of well established or existing drug substances and products[10,11,12]. Even the United States Pharmacopoeia (USP) has a requirement listed under ‘Stability Studies in Manufacturing’, which says that samples of the products should be assayed for potency by the use of a stability-indicating assay [13]. The requirement in such explicit manner is, however, absent in other pharmacopoeias.
Current ICH guideline on Good Manufacturing Practices for Active Pharmaceutical Ingredients (Q7A), which is under adoption by WHO, also clearly mentions that the test procedures used in stability testing should be validated and be stability- indicating [14].
OBJECTIVE OF STABILITY STUDIES
Stability studies are performed to establish the shelf life and storage condition of API and product. In recently adopted stability guidelines, the committee for proprietary medicinal product (CPMP) indicates the objective of stability testing is to provide evidence on how much quality of an API varies with time under influence of the variety of environmental factor such as temperature, humidity and light. The stability of API does not mean “fix” or “not likely change” but it means “controlled and acceptable change”. Force degradation condition, stress agent concentration and time of stress are to be establishing in such a way that, they effect degradation preferably 10-20% of parent constituent. Stability testing is performed for welfare of the patient and to protect reputation of producer, as a requirement of regulatory agencies to provide data that may be of value in formulation of other product [15].
STEPS INVOLVED DURING THE DEVELOPMENT OF STABILITY–INDICATING ANALYTICAL METHODS (SIAMs)
A SIAMs is an estimative analytical method used to detect a trace level amount or residual levels of the API present due to degradation or designing of its synthesis route. As per the FDA regulations, a SIAMs is defined as a completely validated method that accurately and precisely measures API free from potential interferences like degradants, biproducts, intermediates, and exicipients and the FDA recommend that all assay content methodologies for stability studies be stability indicating [16]. There are three components necessary for implementing a SIAMs.
1. Generation of degraded samples for testing selectivity of the method,
2. Method development,
3. Method validation
Step 1: Generation of degraded samples for testing selectivity of the method
Here lies one of the main concerns related to a development of a SIM, since the available guidance documents do not state the extent to which stress tests should be carried out – that is, how much stress should be applied or how much degradation should be aimed for. In fact, there is not a “gold rule” that attends this issue and therefore, it is important to keep in mind that experimental conditions of stress tests, should be realistic and lead to “purposeful degradation” [17].
Stress tests should generate representative samples to assess drug substance and drug product stability, provide information about possible degradation pathway and demonstrate the stability indicating power of the analytical procedures applied.
1) Determination of Limit of Quantification (LOQ)
In close relation to the determination of the amount of degradation is the evaluation of Limit of Detection (LOD) and Limit of Quantification (LOQ) of the method. These limits should be closely related to the Reporting, Identification and Qualification of degradation products, as stated in ICH Q3B (R2) [5].These thresholds aredetermined either as percentage of drug substance or total daily intake (TDI) of degradation product. The analytical methods are usually expected to be validated for the ability to quantify potential degradation products and drug impurities with a LOD and LOQ at least as sensitive as the ICH threshold (see Figure 1).
Figure 1: ICH thresholds for degradation products in New Drug Application (ICH Q3B)
Reporting threshold
Maximum daily dose 1 Threshold 2,3
≤ 1 g 0.1%
> 1 g 0.05%
Identification threshold
Maximum daily dose1 Threshold 2,3
<1mg 1.0% or 5μg TDI, whichever is lower
1mg-10mg 0.5% or 20μg TDI, whichever is lower
>10mg-2g 0.2% or 2mg TDI, whichever is lower 0.10%
Qualification threshold
Maximum daily dose1 Threshold 2,3
<10 mg 1.0% or 50μg TDI, whichever is lower
10mg – 100 mg 0.5% or 200μg TDI, whichever is lower
>100mg-2g 0.2% or 3mg TDI, whichever is lower
>2g 0.15%
Note:
1. The amount of drug substance administered per day
2. Threshold for degradation product are express either as percentage of the drug substance or as total daily intake (TDI) of the degradation product. lower threshold can be appropriate if the degradation product unusually toxic
3. Higher threshold should be scientifically justified.
The identification threshold (IT) varies from 0.1 to 1.0% of the labeled amount of active ingredient in the dosage form, or from 5μg to 2 mg TDI, depending on the maximum daily dosage in the product´s professional labeling. The identification threshold may be lowered for degradation products that may be exceptionally toxic. The Reporting Threshold (RT) is either 0.1% or 0.05% depending on the maxim daily dosage. For very low dose drug products, where this type of sensitivity is not attainable, even after exhaustive tentative, justification may be provided describing the failed reports. Process-related drug substance impurities that are also degradation products should have the same limits as for ICH Q3B. Ideally, the same analytical methodology should be used for Quality Control and Stability Studies. The determination of Out-of-Specification or Out-of-Trend results should be more reliable, when using a SIM, since LOD and LOQ used allows detection of impurities and/or degradation products adequately. In the situation in which a new peak arises during stability study and one may expect that it should not exist and hence it would constitute a type of OOT, the use of a well studied and well determined LOQ in a SIM, will help the applicator to decide if additional action are needed to investigate a new substance or a OOT.
It should be mentioned that these thresholds are established for new drug products or New Drug Application (NDA). For Abbreviated New Drug Application (ANDA) or generic drugs, there are not specific regulations about this topic and even less, the companies dealing with these products, have background information as those obtained in the development of NDA. Such application is expected to contain a “full description of the drug substance including its physical and chemical characteristics and stability as well; such specifications and analytical methods are necessary to assure the identity, strength, quality, purity and bioavailability of drug product and stability data with proposed expiration date”. As already cited, for ANDA, there are not specific regulations and the same ICH recommendation has been used. However, precisely because of the lack of information derived from the new drug development, the complexity and responsibility in developing/validating a SIM for an ANDA is high. Information like aqueous solubility, pH versus solubility profile, excipients compatibility studies, etc, all information that enable fully assume the knowledge of the product ,will help to ensure that best (more appropriate) condition were chosen for developing a SIM, like those related to the forced degradation design.
2) Overstressing / Understressing
Care should be taken in order to avoid overstressing or understressing samples, with may lead to non representative or non-purposeful degradation. So, the use of a properly designed and executed forced degradation study will generate representative samples that will help to ensure that resulting method reflects adequately long-term stability [18]. About the forced degradation (or stress test, both terms will be used in the text) design, it is recommended [19] to include alkaline and acidic hydrolysis, photolysis, oxidation, humidity and temperature stress. An compilation of data from literature [19-23] is shown at Table 1 and compiles the more often used conditions to perform forced degradation studies. These conditions can be used as a starting point in the development of a SIM. Changing conditions to harsher or softer levels, can be applied, when too little or too much degradation are obtained. For example, in cases in which too little degradation was obtained in the hydrolyses stress, it is recommended to increase concentrations to 1 Mol L-1 or higher; for oxidation stress, increase peroxide concentration to 10% or 20% (v/v) and/or time of reaction, as well as temperature. If co-solvents are necessary to increase solubility, it is recommended the use of acetonitrile that does not work as a sensitizer in photostability stress. Data needs to be evaluated as unusual degradants may form with co-solvents. If even not all conditions may cause degradation, document efforts and severity of conditions and should be include in final report. By the other side, if too much degradation is detected, the severity of conditions may be decreased, by diluting acid/bases, neutralizing, reducing exposure time.Also, need to be clarified, that synthesis impurities when are not also degradation products do not need to be described in Stability Studies, but SIM may assure that these impurities do not interfere on degradation products determination.
Table 1: “More often” used conditions for forced degradation studies
| Solid State | |||
| Stress | Condition | Period of time | |
| Heat | 60° C | Up to 1 month | |
| Humidity | 75% RH | Up to 1 month | |
| Photostability | 3 mm (powder)Exposed and non-exposedsamples (“control”) | Follow ICH requirements(Q1B) | |
| Solution State | |||
| Stress | Condition | Period of time | |
| Hydrolysis | Acid | 0.1 – 1 Mol L-1 HCl | Up to weeks and 60° C |
| alkaline | 0.1 – 1 Mol L-1 NaOH | Up to weeks and 60° C | |
| Oxidation | H2O2 3% (v/v) | Up to 24 hours | |
| Photostability | Exposed and non-exposedsamples (“control”) | Follow ICH requirements(Q1B) | |
| Heat | 60° C | Up to 1 month |
3) FORCED DEGRADATION STUDIES (STRESS STUDIES)
Forced degradation or stress studies are undertaken to deliberately degrade the sample. These studies are used to evaluate an analytical method’s ability to measure an active ingredient and its degradation products, without interference, by generating potential degradation products. During validation of the method, drug substance are exposed to acid, base, heat, light and oxidizing agent to produce approximately 10% to 30% degradation of active substance. The studies can also provide information about the degradation pathways and degradation products that could form during storage. These studies may also help in the formulation development, manufacturing, and packaging to improve a drug product. Reasons for carrying out forced degradation studies include: development and validation of stability-indicating methodology, determination of degradation pathways of drug substances and drug products, discernment of degradation products in formulations that are related to drug substances versus those that are related to non–drug substances (e.g., excipients) [17,22].
APPROPRIATE TIMING
It is very important to know when to perform forced degradation studies for the development of new drug substance and new drug product. FDA guidance states that stress testing should be performed in Phase III of regulatory submission process. Stress studies should be done in different pH solutions, in the presence of oxygen and light, and at elevated temperatures and humidity levels to determine the stability of the drug substance. These stress studies are conducted on single batch. The results should be summarized and submitted in an annual report [24]. However, starting stress testing early in preclinical phase or phase I of clinical trials is highly encouraged and should be conducted on drug substance to obtain sufficient time for identification of degradation products and structure elucidation as well as optimizing the stress conditions. An early stress study also gives timely recommendations for making improvements in the manufacturing process and proper selection of stability-indicating analytical procedures [23,25].
LIMITS FOR DEGRADATION
The question of how much degradation is sufficient has been the topic of many discussions amongst pharmaceutical scientists. Degradation of drug substances between 5% to 20% has been accepted as reasonable for validation of chromatographic assays [26,27]. Some pharmaceutical scientists think 10% degradation is optimal for use in analytical validation for small pharmaceutical molecules for which acceptable stability limits of 90% of label claim is common [28]. Others suggested that drug substance spiked with a mixture of known degradation products can be used to challenge the methods employed for monitoring stability of drug product [22]. No such limits for physiochemical changes, loss of activity or degradation during shelf life have been established for individual types or groups of biological products [29].
It is not necessary that forced degradation would result in a degradation product. The study can be terminated if no degradation is seen after drug substance or drug product has been exposed to stress conditions than that conditions mentioned in accelerated stability protocol [30].This is indicative of the stability of the molecule under test. Over-stressing a sample may lead to the formation of secondary degradation product that would not be seen in formal shelf life stability studies and under-stressing may not generate sufficient degradation products [31]. Protocols for generation of product-related degradation may differ for drug substance and drug product due to differences in matrices and concentrations. It is recommended that maximum of 14 days for stress testing in solution (a maximum of 24 h for oxidative tests) to provide stressed samples for methods development [32].
EXPERIMENTAL DESIGN
In designing forced degradation studies, it must be remembered that more strenuous conditions than those used for accelerated studies (25°C/60% RH or 40°C/75% RH) should be used. At a minimum, the following conditions should be investigated:
(1) Acid and base hydrolysis,
(2) Hydrolysis at various ph,
(3) Thermal degradation,
(4) Photolysis, and
(5) Oxidation. For the drug substance and drug product, the scheme shown in Figure 2 could be used as a guide.
The initial experiments should be focused on determining the conditions that degrade the drug by approximately 10%. The conditions generally employed for forced degradation are summarized in Table 2.
However, some scientists have found it practical to begin at extreme conditions (80°C or even higher, 0.5N NaOH, 0.5N HCl, 3% H2O2) and testing at shorter (2, 5, 8, and 24 hrs, etc) multiple time points, thus allowing for a rough evaluation of rates of degradation. Testing at early time points may permit distinction between primary degradants and their secondary degradation products. This strategy allows for better degradation pathway determination. It must be noted that a forced degradation study is a “living process” and should be done along the developmental time line as long as changes in the stability-indicating methods, manufacturing processes, or formulation changes are ongoing.
Forced degradation is only considered complete after the manufacturing process is finalized, formulations established, and test procedures developed and qualified. The conditions listed in Table 1 are by no means exhaustive and should be adjusted by the researcher as needed to generate ~10% degradation of the API.
The nature (inherent stability/instability) of the particular drug substance will determine in which direction to adjust the stress conditions. Also, the aforementioned conditions could be used to stress the drug substance or drug product either in the solid or liquid/suspension form as applicable.
For oxidative degradation with H2O2, at least one of the storage conditions should be at room temperature. Heating H2O2 solution increases the homolytic cleavage of the HO-OH bond to form the alkoxy radical. The alkoxy radical is very reactive and may come to dominate the observed degradation pathway. Adding a small quantity of methanol in a confirmatory stress experiment quenches the alkoxy radical and rules out species produced by this more aggressive oxidizing agent. Also, the formation of peroxycarboxymidic acid has been observed when acetonitrile is used as a cosolvent in H2O2 stress studies (in basic conditions). The peroxycarboximidic acid has activated hydroxylation reactivity, which is not representative of H2O2. To circumvent these problems, some research scientists always perform a parallel or alternative oxidative study using azobisisobutyronitrile (AIBN), which is a less reactive oxidant and has been shown to produce more representative degradants.
List of some common conditions used in conducting forced degradation studies for drug substances as shown in Table 3.

Figure 2: An Illustrative Diagram Showing the Different Forced Degradation Condition to be used for Drug Substance and Drug Product
Table 2: Conditions Generally Employed For Forced Degradation 24
| Degradation Type | Experimental Condition | Storage Condition | Sampling Time |
| Control API (no acid or base) | 40 0C, 600C | 1,3,5 days | |
| Hydrolysis | 0.1N NAOHAcid Control(no API)Base Control(no API)pH: 2,4,6,83% H2O2 | 40 0C, 600C40 0C, 600C40 0C, 600C40 0C, 600C25 0C, 600C | 1,3,5 days1,3,5 days1,3,5 days1,3,5 days1,3,5 days |
| Oxidation | Peroxide controlAzobisisobutyronitrile (AIBN)AIBN Control | 25 0C, 600C40 0C, 600C40 0C, 600C | 1,3,5 days1,3,5 days1,3,5 days |
| Photolytic | Light, 1X ICHLight, 3X ICHLight Control | NANANA | 1,3,5 days1,3,5 days1,3,5 days |
| Thermal | Heat chamberHeat chamberHeat chamberHeat chamberHeat control | 60 0C60 0C /75% RH80 0C80 0C /75% RHRoom Temp. | 1,3,5 days1,3,5 days1,3,5 days1,3,5 days1,3,5 days |
Table 3: lists some common conditions used in conducting forced degradation studies for drug substances
| Sample condition | Time / Exposure |
| Solid / 60 – 70ºCSolid / 60 – 70ºC / 75% RHSolid / simulated sunlight0.1 to 2 N HCl solutions either at RT or at 60 – 70ºC0.1 to 1 N NaOH solutions either at RT or at 60 – 70ºCDilute hydrogen peroxide (0.1 to 6%) at RT or at 60 – 70ºCSolution in Water or at 60 – 70ºC | 7 – 10 days10 days2 – 3 weeks x ICH confirmatory exposure1 – 3 days1 – 3 days1 – 3 days1 – 3 days |
SELECTION OF DRUG CONCENTRATION
Which concentration of drug should be used for degradation study has not been specified in regulatory guidance. It is recommended that the studies should be initiated at a concentration of 1 mg/ml [33]. By using drug concentration of 1 mg/ml, it is usually possible to get even minor decomposition products in the range of detection. It is suggested that some degradation studies should also be done at a concentration which the drug is expected to be present in the final formulations [34]. The reason for proposing this are the examples of aminopenicillins and aminocephalosporins where a range of polymeric products have been found to be formed in commercial preparations of containing drug in high concentrations [35].
Example-The goal is to degrade the active moiety to 5-10% in sample for which the conditions can be used as Shown in Table 4.
Table 4: Conditions for different pH
| Study | Conditions |
| Acidic pH | 0.1N HCl |
| Neutral pH | pH 7.0 phosphatebuffer |
| Basic pH | 0.1N NaOH |
| Oxidation | O2 Atmosphere, H2O2 |
| Photolysis (UV) | 1000 Watts Hrs/M2 |
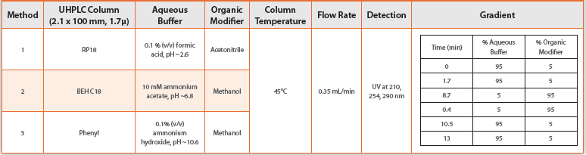
| Photolysis(Fluorescence)METHOD DEVELOPMENT AND OPTIMIZATION Before starting method development, various physiochemical properties like pKa value, log P, solubility and absorption maximum of the drug must be known for it lays a foundation for HPLC method development. Log P and solubility helps select mobile phase and sample solvent while pKa value helps determine the pH of the mobile phase [34]. Reverse phase column is a preferred choice to start the separation of sample components as the degradation is carried out in aqueous solution. Methanol, water and acetonitrile can be used as mobile phase in various ratios for the initial stages of separation. Selection between methanol and acetonitrile for organic phase is based on the solubility of the analyte. Initially the water: organic phase ratio can be kept at 50: 50 and suitable modifications can be made as trials proceed to obtain a good separation of peaks. Latter buffer can be added if it is required to obtain better peak separation and peak symmetry. If the method is to be extended to LC-MS then mobile phase buffer should be MS compatible like triflouroacetic acid and ammonium formate. Variation in column temperature affects the selectivity of the method as analytes respond differently to temperature changes. A temperature in the range of 30-40°C is suitable to obtain good reproducibility [16].It is better to push the drug peak farther in chromatogram as it results in separation of all degradation products. Also a sufficient run time after the drug peak is to be allowed to obtain the degradants peak eluting after the drug peak [33]. During method development it may happen that the drug peak may hide an impurity or degradant peak that co-elutes with the drug. This requires peak purity analysis which determines the specificity of the method. Direct analysis can be done on line by using photo diode array (PDA) detection. PDA provides information of the homogeneity of the spectral peak but it is not applicable for the degradants that have the similar UV spectrum to the drug. Indirect method involves change in the chromatographic conditions like mobile phase ratio, column, etc. which will affect the peak separation. The spectrum of altered chromatographic condition is then compared with the original spectra. If the degradant peaks and area percentage of the drug peak remains same then it can be confirmed that the drug peak is homogeneous [36]. The degradant that co-elutes with the drug would be acceptable if it is not found to be formed in accelerated and long term storage conditions [1]. The method is then optimized for separating closely eluting peaks by changing flow rate, injection volume, column type and mobile phase ratio.METHOD VALIDATION The developed SIM is then validated according to USP/ICH guideline for linearity, accuracy, precision, specificity, quantitation limit, detection limit, ruggedness and robustness of the method. It is required to isolate, identify and quantitate the degradants found to be above identification threshold (usually 0.1%)[37,38]. If the method does not fall within the acceptance criteria for validation, the method is modified and revalidated [36].Validation is defined by the International Organization for Standardization (ISO) as “verification, where the specified requirements are adequate for an intended use”, where the term verification is defined as “provision of objective evidence that a given item fulfills specified requirements” [39]. The applicability and scope of an analytical method should be defined before starting the validation process. It includes defining the analytes, concentration range, description of equipment and procedures, validation level and criteria required. The validated range is defined by IUPAC as “the interval of analyte concentration within which the method can be regarded as validated” [40,41]. This range does not have to be the highest and lowest possible levels of the analyte that can be determined by the method. Instead, it is defined on the basis of the intended purpose of the method [42,43]. The method can be validated for use as a screening (qualitative), semi-quantitative (e.g. 5-10ppm) or quantitative method. It can also be validated for use on single equipment, different equipments in the laboratory, different laboratories or even for international use at different climatic and environmental conditions. The criteria of each type of validation will of course be different with the validation level required[39,44].APPROACHES TO VALIDATION EXPERIMENTS Accuracy is established by quantitation of the sample against a reference standard for API, or spiking placebo with API for drug product. It can also be determined by comparison of results from alternate measurement techniques.Precision is determined by multiple measurements on an authentic, homogeneous set of samples. Samples may be analyzed on different days, by different analysts, on different instruments, or in different laboratories. There are three levels of precision validation evaluations – repeatability, intermediate precision, and reproducibility. Repeatability is a measure of precision under the same conditions over a short period of time. Intermediate precision is a measure of precision within the same laboratory by different operators, using different instruments, and making measurements on different days. Reproducibility assesses precision between two or more laboratories.Specificity can be established by a number of approaches, depending on the intended purpose of the method. The ability of the method to assess the analyte of interest in a drug product is determined by a check for interference by placebo. Specificity can be assessed by measurement of the API in samples that are spiked with impurities or degradants, if available. If API-related compounds are not available, drug can be stressed or force-degraded in order to produce degradation products. In chromatographic separations, apparent separation of degradants may be confirmed by peak purity determinations by photodiode array, mass purity determinations by mass spectroscopy (MS), or by confirming separation efficiency using alternate column chemistry. During forced degradation experiments, degradation is targeted at 5 to 20% degradation of the API, in order to avoid concerns about secondary degradation.The limit of Detection and limit of Quantitation are based on measurement Signal-to-noise ratios of 3 and 10, respectively. Standards or samples at concentrations near the expected limits are measured. Signal- to-noise can be generated by software, manually measured, estimated from standard deviation calculations, or limits may be empirically determined.Linearity is established by measuring response at various concentrations by a regression plot, typically by method of least squares. The response may require mathematical manipulation prior to linearity assessments. A visual inspection of the linearity plot is the best tool for examining proportionality of the response. The range is established by the required limits of the method and the point at which linearity is compromised.Robustness is typically assessed by the effect of small deliberate changes to chromatographic methods on system suitability parameters such as peak retention, resolution, and efficiency. Experimental factors that are typically varied during method robustness evaluations include[45]: (i) Age of standards and sample preparations, (ii) Sample extraction time, (iii) Variations to pH of mobile phase, (iv) Variation in mobile phase composition, (v) Analysis temperature, (vi) Flow rate, (vii) Column lot and/or manufacturer, (viii) Type and use of filter against centrifugation. Robustness experiments are an ideal opportunity to utilize statistical design of experiments, providing data driven method control.The ICH guidance on validation separates types of methods according to the purpose of the method and lists which evaluations are appropriate for each type [28].The ICH guidance also suggests detailed validation schemes relative to the intended purpose of the methods. It lists recommended data to report for each validation parameter. Acceptance criteria for validation elements must be based on the historical performance of the method, the product specifications, and must be appropriate for the phase of drug development.OTHER ANALYTICAL METHODS FOR DEVELOPING SIM Stability-indicating methods will be characterized by potency, purity and biological activity[46]. The selection of tests is product specific. Stability indicating methods may include various methods like electrophoresis (SDS-PAGE, immunoelectrophoresis, Western blot, isoelectrofoccusing), high-resolution chromatography (e.g., reversed phase chromatography, SEC, gel filtration, ion exchange, and affinity chromatography) and peptide mapping[47]. The analytical method of choice should be sensitive enough to detect impurities at low levels (i.e., 0.05% of the analyte of interest or lower) and the peak responses should fall within the range of detector’s linearity. The analytical method should be capable of capturing all the impurities formed during a formal stability study at or below ICH threshold limits[48,49]. Degradation product identification and characterization are to be performed based on formal stability results in accordance with ICH requirements. Conventional methods (e.g., column chromatography) or hyphenated techniques (e.g., LC–MS, LC–NMR) can be used in the identification and characterization of the degradation products. Use of these techniques can provide better insight into the structure of the impurities that could add to the knowledge space of potential structural alerts for genotoxicity and the control of such impurities with tighter limits[37,47,50]. It should be noted that structural characterization of degradation products is necessary for those impurities formed during formal shelf-life stability studies and above the qualification threshold limit[48].New analytical technologies that are continuously being developed can also be used when it is appropriate to develop stability indicating method[51]. The unknown impurity, which is observed during the analysis, pharmaceutical development, stress studies and formal stability studies of the drug substances and drug product, can be separated and analyzed by using various chromatographic techniques like Reversed Phase High Performance Liquid Chromatography (RP-HPLC), Thin Layer Chromatography (TLC), Gas Chromatography (GC), Capillary Electrophoresis (CE), Capillary Electrophoresis Chromatography (CEC) and Super critical Fluid Chromatography (SFC). An excellent combination of hyphenated chromatographic and spectroscopic technique such as HPLC-DAD (High Performance Liquid Chromatography Photodiode Array ultraviolet Detector), LC-MS (Liquid Chromatography-Mass Spectrometry), LC-NMR (Liquid Chromatography-Nuclear Magnetic Resonance) and GCMS (Gas Chromatography-Mass Spectrometry) are used when degradants cannot be isolated in pure form. HPLC-DAD and LC-MS are used to compare the RRT (relative retention time), UV spectra, mass spectra (MS/MS or MSN)[52]. Ranjit Singh et al. discussed the role of hyphenated systems for the isolation of degradants and impurities[50].CONCLUSION Stability-indicating method is an analytical procedure that is capable of discriminating between the major active (intact) pharmaceutical ingredients (API) from any degradation (decomposition) product(s) formed under defined storage conditions during the stability evaluation period.Forced degradation studies are indispensable in the development of stability-indicating and degradant-monitoring methods as part of a validation protocol. Forced degradation studies also provide invaluable insight in investigating degradation products the use of properly designed and executed forced degradation study will generate a representative sample that will in turn help to develop stability-indicating method.REFERENCES 1.International Conference on Harmonization (ICH) (2003) Harmonised Tripartite Guideline on, Topic Q1A, Notes for Guidance on stability testing: Stability testing of new drug substances and new drug products products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 2.Swartz M, Krull I (2004) Investigating Out-Of-Specifications Results. LCGC 22: 132-136. 3.Center for Drug Evaluation and Research (CDER) (2006) Guidance for Industry Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production. 4. International Conference on Harmonization (ICH) (1993) Harmonised Tripartite Guideline Notes for Stability Testing of New Drug Substances and Products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 5.International Conference on Harmonization (ICH) (1996) Harmonised Tripartite Guideline Notes for Impurities in New Drug Products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 6.International Conference on Harmonization (ICH) (1999) Harmonised Tripartite Guideline Notes for Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 7.International Conference on Harmonization (ICH) (1995) Harmonised Tripartite Guideline Notes for Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 8.FDA (1987) Guidance for Industry: Submitting Documentation for the Stability of Human Drugs and Biologics. Food and Drug Administration, Rockville, MD. 9.FDA (1998) Guidance for Industry: Stability Testing of Drug Substances and Drug Products (Draft guidance), Food and Drug Administration, Rockville, MD. 10. WHO (1996) Guidance for Stability Testing of Pharmaceutical Products Containing Well Established Drug Substances in Conventional Dosage Forms, in WHO Expert Committee on Specifications for Pharmaceutical Preparations. Technical Report Series 863, World Health Organization, Geneva, 65–79. 11.CPMP (1998) Guidance on Stability Testing of Existing Active Substances and Related Finished Products. Committee for Proprietary Medicinal Products, EMEA, London. 12.TPD (1997) Stability Testing of Existing Drug Substances and Products. Therapeutic Products Directorate, Ottawa. 13. United States Pharmacopeia 24(2000) Rockville, MD: USP Convention Inc. 14.International Conference on Harmonization (ICH) (2000) Harmonised Tripartite Guideline Notes for Good Manufacturing Practices for Active Pharmaceutical Ingredients. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 15.Sehrawat R, Maithani M, Singh R, Regulatory aspects in development of stability indicating method: A Review. Chromatographia 2010; 72: 1-6. 16.Snyder L.R., Glajch J.L., Kirkland J.J., Practical HPLC method Development. New York: John Wiley 1988; 227-251 17.Ngwa G, Forced Degradation Studies. Forced Degradation as an Integral part of HPLC Stability Indicating Method Development Drug Delivery Technology 2010. 18. International Conference on Harmonization (ICH) (2003) Harmonised Tripartite Guideline on, Topic Q1A (R2), Notes for Guidance on Stability Testing of new Drug Substances and Product. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 19.Klick S, Muijselaar P, Waterval J, Eichinger T, Korn C, Toward a Generic Approach for Stress Testing of Drug Substances and Drug Products. Pharm. Technol 2005; 29: 48-57. 20.Alsante K.M., Ando A, Brown R, Ensing J, Hatajik T.D., The Role of Degradant Profiling in Active Pharmaceutical Ingredients and Drug Products. Adv. Drug Delivery Rev 2007; 59:29—37. 21.Reynolds D, Forced Degradation of Pharmaceuticals. Am. Pharm 2004; 56-61. 22.Reynolds D, Facchine K, Mullaney F, Alsante K, Hatajik T, Available Guidance and Best Practices for Conducting Forced Degradation Studies. Pharmaceutical Technology 2002; 48-56. 23.Kats M, International Forced Degradation Studies: Regulatory Considerations and Implementation 2005; 18:1-7. 24.FDA (2003) Guidance for Industry: INDs for Phase II and III Studies – Chemistry, Manufacturing, and Controls Information, Food and Drug Administration. Rockville, MD. 25..FDA (1999) Guidance for Industry: INDs for Phase 2 and 3 Studies of Drugs, Including Specified Therapeutic Biotechnology-Derived Products, Draft Guidance, Food and Drug Administration. Rockville, MD. 26.Szepesi G, Selection of high-performance liquid chromatographic methods in pharmaceutical analysis. J. Chromatogr 1989; 464: 265-278. 27.Carr G.P., Wahlich J.C., A practical approach to method validation in pharmaceutical analysis. J. Pharm. Biomed. Anal 1990; 86: 613-618. 28.Jenke D.R., Chromatographic method validation: A review of common practices and procedures II. J. Liq. Chromatogr 1996; 19: 737-757. 29.International Conference on Harmonization (ICH) (1996) Harmonised Tripartite Guideline on Stability Testing of Biotechnological/Biological Products Availability. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 30.International Conference on Harmonization (ICH) (1996) Harmonised Tripartite Guideline on, Topic Q1B, Notes for Guidance on Photo stability Testing of New Drug Substances and Product. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 31.Maheswaran R, FDA perspectives: scientific considerations of forced degradation studies in ANDA submissions. Pharm. Technol 2012; 36: 73-80. 32.Klick S, Pim G.M., Waterval J, Thomas E, Alexander J, Toward a generic approach for stress testing of drug substances and drug products. Pharm. Technol 2005; 29:48–66. 33.Bakshi M, Singh S, Development of validated stability-indicating assay methods-critical review. J. Pharm. Biomed. Anal 2002; 28: 1011-1040. 34.Singh S, Bakshi M, Guidance on conduct of stress tests to determine inherent stability of drugs. Pharm. Technol 2000; 24:1-14. 35.Larsen C, Bundgaard H, Polymerization of Penicillins.V. Separation, identification and quantitative determination of Antigenic Polymerization products in Ampicillin Sodium preparations by high-performance liquid chromatography. J. Chromatogr 1978; 147: 143–150. 36.Riddhiben M.P., Piyushbhai M.P., Natubhai M.P., Stability indicating HPLC method development – a review. Int. Res. J. Pharm 2011; 2:79-87. 37.International Conference on Harmonization (ICH) (2000) Harmonised Tripartite Guideline Notes for Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 38.Maha M.A.R., Alia N.W., Sayed S.A., Sayed H.Z., Mohamed A, Validated stability indicating methods for determination of Nitazoxanide in presence of its degradation products. J. Pharm. Anal 2012; 2:105-116. 39.ISO/IEC (2007) (1stedn) Guidance on International vocabulary of metrology – Basic and general concepts and associated terms (VIM). International Organization for Standardization. Geneva. 40.Perlatti B, Maria F.D., Gracas F.D.S., Fernandes J.B., Ross M, Validation and application of HPLC–ESI-MS/MS method for the quantification of RBBR decolorization, a model for highly toxic molecules, using several fungi strains. Bio Tech 2012; 124:37–44. 41.Thompson M, Ellison S.L.R., Wood R, Harmonized guidelines for single-laboratory validation of methods of analysis – IUPAC technical report. Pure. Appl. Chem 2002; 74:835-855. 42.Susan D, Van A, Henry J.N., Chinfong C, HPLC method validation studies on a specific assay for monomethoxypoly (ethylene glycol) succinimido carbonate (mPEG-SC). J Pharmaceut Biomed 2009; 50:138–143. 43.Potts A.R., Tatiana P, Cassandra J, Luba P, Ahalya W, Validation of a quantitative HPLC method for bacitracin and bacitracin zinc using EDTA as a mobile-phase modifier. J Pharmaceut Biomed Anal 2012; 70:619–623. 44.Hill A.R.C., Reynolds S.L., Guidelines for in-house validation of analytical methods for pesticide residues in food and animal feeds. Analyst 1999; 124:953-958. 45.International Conference on Harmonization (ICH) (2005) Harmonised Tripartite Guideline on, Topic Q2 (R1), Notes for Guidance on Validation of Analytical Procedures, Text and Methodology. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 46.Bichsel V.E., Curcio V, Gassmann R, Otto H, Requirements for the quality control of chemically synthesized peptides and biotechnologically produced proteins. Pharm. Acta Helv 1996; 71: 439-446. 47.EMA (2007) Guideline on the Limits of Genotoxic Impurities, Committee for Medical Products for Human Use (CHMP). 48.International Conference on Harmonization (ICH) (2006) Harmonised Tripartite Guideline on, Topic Q3A (R2), Notes for Guidance on Impurities in New Drug Substances. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 49.International Conference on Harmonization (ICH) (2006) Harmonised Tripartite Guideline on, Topic Q3B (R2), Notes for Guidance on Impurities in New Drug Products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 50.Singh R, Rehman Z, Current trends in forced degradation study for pharmaceutical product development. J. Pharm. Educ. Res 2012; 3: 54-63. 51.International Conference on Harmonization (ICH) (1999) Harmonised Tripartite Guideline on, Topic Q6B, Notes for Guidance on Specifications: Test Procedures and Acceptance Criteria for Bio-technological/Biological Products. Pub by The European Agency for the Evaluation of Medicinal Products, Human medicine evaluation Unit. 52.Qiu F, Norwood D.L., Identification of pharmaceutical impurities. J. Liq. Chromatogr 2007; 30:877-935. | 6 x 106 Lux Hrs |