

Abstract
Current standards and regulations demand the pharmaceutical industry not only to produce highly pure drug substances, but to achieve a thorough understanding of the impurities accompanying their manufactured drug substances and products. These challenges have become important goals of process chemistry and have steadily stimulated the search of impurities after accelerated or forced degradation procedures.
As a result, impurity profiling is one of the most attractive, active and relevant fields of modern pharmaceutical analysis. This activity includes the identification, structural elucidation and quantitative determination of impurities and degradation products in bulk drugs and their pharmaceutical formulations.
Nuclear magnetic resonance (NMR) spectroscopy has evolved into an irreplaceable approach for pharmaceutical quality assessment, currently playing a critical role in unequivocal structure identification as well as structural confirmation (qualitative detection), enabling the understanding of the underlying mechanisms of the formation of process and/or degradation impurities.
NMR is able to provide qualitative information without the need of standards of the unknown compounds and multiple components can be quantified in a complex sample without previous separation. When coupled to separative techniques, the resulting hyphenated methodologies enhance the analytical power of this spectroscopy to previously unknown levels. As a result, and by enabling the implementation of rational decisions regarding the identity and level of impurities, NMR contributes to the goal of making better and safer medicines.
Herein are discussed the applications of NMR spectroscopy and its hyphenated derivate techniques to the study of a wide range pharmaceutical impurities. Details on the advantages and disadvantages of the methodology and well as specific challenges with regards to the different analytical problems are also presented.
Graphical abstract
Keywords
- NMR spectroscopy;
- Pharmaceutical impurities;
- LC–NMR;
- Structural elucidation;
- Qualitative and quantitative analysis
1. Introduction
1.1. Pharmaceutical impurities and approaches to their modern quality control
The quality assurance and quality control of active pharmaceutical ingredients (APIs) and excipients are major issues in pharmaceutical analysis, which intend to prevent damages to the patients. Quality control methods are regulated in the pharmacopeias and other documents, which are continuously revised in order to keep updated drugs, excipients, and also their methods for analysis. Furthermore, internationally agreed recommendations, such as those issued by the International Conference on Harmonization (ICH) are steadily moving pharmaceutical analysis beyond compendial requirements, with the advantage of the most modern analytical technologies [1]. Pharmaceutical impurities are the unwanted chemicals that remain with the API, develop during formulation, or upon degradation of both API and drug products. Their presence, even in small amounts, might influence the efficacy and safety of the pharmaceutical products; therefore, drug purity has always been associated to drug quality.
The evolution of chemical knowledge and the advent of increasingly sensitive and selective analytical methods have been continuously stimulating the interest in the determination of drug purity and the impurities themselves in both, natural and synthetic products. This is in line with the policy of the pharmaceutical industry, which has always demanded that the API should be as pure as possible. The number of recently published books [2], book chapters [3], [4], [5] and [6] and papers on the subject demonstrate the increasing importance of pharmaceutical impurities [7], [8], [9], [10] and [11] and impurity profiling [12], [13], [14], [15] and [16]. These and other [17], [18] and [19] publications also reflect the continued interest of the regulatory authorities, industry and scientists in these areas.
The ICH Q3A guideline states that impurity profile is “a description of the identified and unidentified impurities, present in a new drug substance” [20]. According to the guide, the impurities are classified into organic, inorganic and organic volatile impurities. The organic impurities can arise from the manufacturing process and/or be formed during storage of the drug substances. They include starting materials, by-products, synthetic intermediates, degradation products, reagents, ligands and catalysts. As specifically indicated, the guide does not cover enantiomeric impurities and polymorphic forms.
The ICH Q3B guideline addresses impurities in new drug products, classified as degradation products of the drug substance or reaction (interaction) products of the drug substance with an excipient and/or immediate container closure system [21].
In addition, in current pharmaceutical development processes, impurities in the API have identification and qualification thresholds of 0.10 and 0.15% respectively, for doses of the API up to 2 g/day [20] and many recent publications reflect interest in compounds ranging from 0.01 to 0.1%.
Reliable assessment of drug purity in accordance with these stringent standards requires the use of state of the art validated analytical methods, increasingly sensitive detections, and mainly an analyst prepared to critically interpret the results.
The available tools for impurity profiling have already been reviewed [22]. Reversed-phase liquid chromatography (RP–LC) is still the primary method for analyte separation toward impurity profiling of drugs. However, an increasing number of examples show the usefulness of both nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) as additional tools for the detection and quantitation of difficult to solve impurity challenges.
When employed as stand-alone techniques, NMR and MS offer a high degree of orthogonality and complementarity [23]. In particular, NMR spectroscopy is relevant because it can simultaneously and selectively detect multiple components in a sample, even without their physical separation.
Mass spectrometric studies can provide structure-rich information of organic compounds, even those found at trace impurity levels, with small amounts of sample. When hyphenated with an LC separation, and when electro-spray (ESI) or atmospheric-pressure chemical ionization (APCI) are used, MS provides very important information about the molecular weights (especially when implemented in high-resolution mode) of the chromatographed compounds. It can also provide the structure (through the fragmentation pattern) of the analytes, especially when some prior information on the molecular skeleton is available and if only minor changes are present in the impurity with respect to the drug substance [24].
Therefore LC–ESI–MS and LC–ESI–MS/MS are currently well-established analytical tools in the rapid identification and characterization of each component in sample mixtures. However, the resulting picture is still often limited with respect to details of molecular structure. Mass spectral methods are rarely capable of de-novo structure determination with full certainty; furthermore, they do not always provide unambiguous structural information and may not be able to distinguish between isomers [25].
In these cases, the next and often final stage is to resort to NMR spectroscopy (usually off-line, but in some cases on-line), in order to access the required information. Occasionally, single crystal X-ray diffractometry and/or synthesis of the proposed structures have been used as final proofs of the impurities’ structure.
Furthermore, NMR is at the center of the new complete molecular confidence concept, which tends to exploit its synergy with LC–MS in order to obtain an estimation of the purity, concentration, and identity of chemical compounds [26].
1.2. NMR spectroscopy
1.2.1. NMR spectroscopy as an analytical tool: Advantages and disadvantages
NMR spectroscopy, an information rich analytical tool, is capable to deliver the most complete molecular structural information, including atomic connectivity, spatial geometry, conformation, and stereochemistry.1H is the most simple and sensitive, and therefore most frequently detected nucleus. Many more isotopes of elements in the periodic table (11B, 13C, 15N, 17O, 19F, 31P, etc.) are also accessible by NMR. In 1H NMR spectroscopy, 1H chemical shifts and 1H–1H couplings are selectively detected, which relate to the chemical environment of the observed nucleus. The spectral signature of each compound is unique, turning the method highly specific for chemical structures and ideally suited for structural elucidation and confirmation.
Since, the intensity of any given NMR signal depends only on the total amount of the detected nuclei in the sample volume (primary ratio rule), quantitative NMR spectroscopy (qNMR) can be used either as an absolute “standard-free” method or as a relative method [27].
The physical principles of the NMR phenomenon and the relevant spectroscopic parameters, such as chemical shift, signal multiplicity, spin–spin coupling constant (J), and the integral intensity, as well as the wide array of modern NMR experiments [28] and [29] are detailed in a number of useful books [30], [31],[32] and [33]. A short list of the most common NMR experiments and the structural information they are able to provide is found in Table 1.
Table 1.Summary of the most relevant 1H and 13C NMR experiments for studying small organic molecules.
| NMR experiment | Acronyma | Information provided |
| 1H NMR | 1D-1H (1H) | Number and types of protons. |
| Nuclear Overhausser effect | 1D-NOE (NOE) | Through space H–H vicinity. |
| Diffusion NMR | DOSY | Molecular size; host–guest interactions. |
| 13C NMR | 1D-13C (13C) | Types of carbons. |
| Special 1D 13C NMR | APTDEPT | Type of carbons (CH, CH2, CH3), according to the number of protons attached to them. |
| 2D Homonuclear (1H–1H) correlation | COSY | Through bond H–H correlations (coupled protons). |
| TOCSY | Through bond long range correlations (coupled spin network). | |
| NOESY | Through space H–H correlations. | |
| ROESY | Long range, through space H–H correlations. | |
| 2D Heteronuclear (1H–13C) correlation | HMBC | Short range (1J) H–C correlations. |
| HSQC | Long range (usually 2J and 3J) H–C correlations. |
a
For a list with the meaning of the acronyms used in this table and throughout the text, see Appendix A, at the end of the manuscript.
NMR spectroscopy has several advantages; it is a robust and quasi-universal detector, has a non-destructive nature, allows easy sample preparation, and simple method development. Although NMR spectroscopy is used in drug impurity profiling mainly after off-line or on-line separation of the impurities, this technique can be a useful tool even without full analyte separation. Spectra are highly specific, this spectroscopy has the ability to provide global information about the sample in a single analysis and it offers multiple calibration options.
Therefore, 1H NMR can be advantageously used as an alternative to conventional high-performance liquid chromatography (HPLC) when availability of impurity standards is not possible, such as during early stages of drug development [34]. This is documented by a steady increase in the volume of reports that employ NMR spectroscopy to solve this problem, but also by the remarkable interest of drug manufacturers in this technique. Several reviews have been published on the application of NMR in pharmaceutical analysis[35] and [36].
Since 1H NMR is sensitive to compounds bearing protons, it can be regarded as a quasi-universal detector, especially for low molecular weight organic molecules. Protons are detected with the same sensitivity, regardless of their chemical environment; thus, the requirement of determining response factors is avoided.
Therefore, NMR ranks among the most informative methods, being a key analytical tool for identification, authentication, detailed structural analysis and elucidation of organic compounds (including stereochemistry and even dynamic effects), and quantitation of unknown natural and synthetic compounds in their mixtures in a single run, provided their signals are well separated [37].
On the other side, the two main disadvantages of NMR are its high equipment cost and its comparatively low sensitivity. When related to MS [Limit of detection (LOD) in the picomolar (pM) range], NMR has lower sensitivity, with LOD values in the low mM range. However, the detection sensitivity of NMR spectroscopy may be enhanced by increasing the number of scans, the concentration of the sample solution and by employing higher magnetic field strengths or modern probes. Accordingly, recent improvements in NMR instrumentation and technology, such as shielded and increasingly stronger high-field magnets [38], cryogenic probes [39] and [40], operation with large sample volumes (several ml), solvent suppression techniques, advanced data processing, versatile pulse sequences and polarization transfer techniques[41], have increased the sensitivity up to the nanomolar range. Other improvements include small OD tubes, Shigemi tubes and microcoil technology.
These characteristics have turned NMR spectroscopy into a powerful tool that can be used in pharmaceutical analysis [42] for the quality control of APIs and excipients for identification, impurity assessment, including adulterations [43] and [44] and assay purposes. Examination of the most recognized Pharmacopeias and an inspection of the recently summarized major compendial applications of NMR spectroscopy in current pharmaceutical analysis [45], reflect an increasingly important participation of this technique in the quality control monographs.
1.2.2. NMR spectroscopy and qualitative pharmaceutical analysis
The power of NMR for structural elucidation is well established; therefore, one of the major applications of NMR experiments is the ‘de novo’ structural elucidation of small organic molecules. The latter is often considered an almost mechanical process, which can be solved mainly with the aid of NMR and/or MS methodologies, especially when the amount of sample is enough to run the powerful two-dimensional NMR (2D-NMR) experiments.
In practice, however, in the case of impurities this represents a rather intellectually and technically challenging problem, because their amounts are often scarce [46], [47], [48] and [49] and their structure is not always related directly to the API. Structural elucidation of trace impurities without separation poses additional demands, requiring minimum signal overlapping between structurally useful spectral signals of the impurity and the resonances of the main component.
However, the information provided by NMR is of such magnitude that the technique can be even used to determine the origin (production plant) of a drug based on its spectral profile [44].
1.2.3. NMR spectroscopy and quantitative pharmaceutical analysis: qNMR
The basic principles of quantitative NMR (qNMR) have been discussed elsewhere [50]. The main advantage of NMR spectroscopy is the linear relationship between the integrated intensities of the resonances from separate components in the spectrum and their molar contents in the sample [27]. The quantitative applications (1H qNMR, qHNMR) are based on the nearly equal response of protons, regardless of their chemical shifts or coupling to other nuclei.
Absolute quantification is possible by relating the peak area of interest in the sample to a signal from an appropriate internal standard, without the need for a reference standard of the same chemical structure as the sample [51] and [52]. When NIST (National Institute of Standards and Technology) standards are used, traceability to the International System of Units (SI) can be established. Relative quantification can be carried out by relating the intensity of the peak of interest to one in the main compound.
In addition, NMR spectroscopy can be considered a relative primary analytical method, because it can be described completely by mathematical equations from which a full uncertainty budget may be derived[53] and [54]. A validation scheme for qNMR methodologies has been proposed [55].
The technique offers a valuable, un-biased and near-universal approach for the analysis of complex mixtures and the simultaneous quantitation of their components, with minimum sample preparation and without the need of a previous separation. Therefore, qHNMR spectroscopy is highly regarded for use in purity assessment of small organic molecules, complementing the identification of potential impurities [56],[57] and [58]. Currently, sensitivity and accuracy of the quantitative results are suitable for controlling low level impurities and qHNMR measurements are at least as reliable and precise as those obtained by chromatographic techniques [59].
Therefore, qNMR is increasingly gaining interest in pharmaceutical applications and has become an essential tool that may be used for establishing sample composition and components’ molar ratio, as well as for assigning content, including the determination of the level of impurities [49]. Furthermore, the capability to perform concurrent identification and quantitation of impurities turns qHNMR into a unique analytical tool. However, the most challenging item is to find proper conditions and separated, pure signals, especially during impurity profiling [60].
An advantage of NMR is that quantitation can be achieved using the main sample component signals thus alleviating the need for time-consuming system suitability and standard runs prior to sample analysis [61]. This strong point was stressed by Deubner and Holzgrabe during their analysis of complex aminoglycoside antibiotics by micellar electrokinetic chromatography, RP–LC and NMR. In that study the authors described the difficulty of evaluating the impurities in gentamicin sulfate which contains no chromophore and has many structurally similar aminoglycoside impurities.
1.2.4. Hyphenated NMR techniques
LC–NMR is one of the most powerful techniques for the characterization of complex mixtures, since it combines a very efficient separation technique and the most important structure elucidation method. However, peak isolation can be also performed by liquid or gas chromatography (GC), as well as by other methods such as capillary electrophoresis (CE). These methods are very different, particularly with respect to sensitivity.
Only recently has LC–NMR coupling become efficient enough and hyphenation allowed the long-expected gain of maximal on-line information about individual constituents of a mixture, with due speed and efficiency. Details of the instrumentation and potential are given in many specific books [62], [63],[64] and [65]; its general applications in pharmaceutical analysis have also been detailed [66].
Analogous hyphenations [67] such as GC–NMR [68] and [69], CZE–NMR [70], [71] and [72], CEC–NMR[73] and [74] and CE–NMR [75], [76], [77] and [78] have also been explored. However, in the on-line mode none of them have been used to solve problems in the area of pharmaceutical impurities and currently this application seems still unfeasible, mainly due to sensitivity problems. To date, CE–NMR is not a well-established method; however, development of smaller volume NMR probes and continuous enhancements of NMR sensitivity will probably transform CE–NMR in the near future into a valuable technique for biopharmaceutical analysis. On the other hand, despite its years of evolution and recent advances, GC–NMR is still in its infancy [79]; even more, its sensitivity, interfacing and other instrumental details require strong improvements.
In its different modes (on-flow, stopped flow and loop-storage techniques) HPLC–NMR may allow unambiguous on-line identification of even very complex structures. An integrated LC–NMR and LC–MS approach toward structural elucidation of impurities without their isolation has been informed [80]. However, very often it is learned that subsequent to having spent a considerable time on separation method development, the amount of structural information collected is limited, because it relates to the amount of time the sample remains in the flow cell. Fraction collection is of help, but attempts to isolate an impurity for off-line structure investigation does not always yield pure substances; instead, the resulting mixtures many times are not easily amenable to analysis, or the quantities are so small that they hinder running 2D NMR experiments.
A recent extension capable of providing NMR data of higher quality includes the insertion of a solid-phase extraction (SPE) unit between the HPLC column and the NMR probe [81], which was also employed for detecting adulterants in an herbal medicine [82]. This HPLC–SPE–NMR setup, where lipophilic stationary phases (silica-C18, styrene-divinylbenzene) are commonly utilized as SPE materials, allows the on-line trapping of the impurity peaks while removing the HPLC eluent.
Use of NMR solvents of high elution power (CD3CN, CD3OD) allows matching the peak volume to the active volume of the NMR flow cell, when the impurities are automatically back-flushed, submitting them to the NMR probe; this results in an overall increase of the sensitivity of the measurement due to analyte accumulation by multiple SPE collections. The overall process requires careful optimization of the SPE trapping and elution conditions, and it must also take into account that use of lipophilic stationary phases may result in loss of the more polar impurities, that cannot be trapped by the SPE stationary phase.
The problem of isolating the impurity for conventional NMR analysis (∼1 mg) can be greatly alleviated by using cryogenically cooled probes, which offer an approximately fourfold increase in S/N, reducing the amount of impurity needing to be purified. Flow and non-flow alternatives are possible, as well as combination with SPE [83]. However, in many cases this does not alleviate the burden of manual preparative-scale isolation of the impurities.
The use of LC–NMR for identification of drug impurities has been impulse during the last 20 years [84]. In this way, NMR and particularly LC–NMR analysis provide complementary information to MS for rapid and unequivocal structural determination. This feature and the appearance of accessible and more powerful spectrometers designed for routine measurements and by the simplicity of the measurements themselves explain the high and increasing interest in NMR spectroscopy on the part of pharmaceutical analysts [46].
2. NMR-assisted Identification/structure elucidation and quantitation of related impurities
According to the ICH Q3A Guideline [20], a “potential impurity is an impurity that theoretically can arise during manufacture or storage”. It has been pointed out that NMR spectrometry is finding increasing application for the identification of unknown organic impurities in pharmaceutical preparations [85]. Impurity profiling is a typical team effort which involves many types of analytical chemists, including UV, IR and NMR spectroscopists, mass spectrometrists, TLC, HPLC and GC chromatographers, as well as experts in hyphenated and several other techniques. When NMR spectroscopy is also necessary for the structure elucidation [86], somewhat larger sample size is necessary. The use of impurity-enriched mother liquors or crude reaction products can overcome problems associated with sensitivity and may be adopted as a general strategy for their identification [87]. These samples are usually obtained by semi-preparative HPLC [88] or by column chromatography.
2.1. Process-related impurities
2.1.1. Impurities resulting from isolation in naturally occurring APIs
Echinochrome is a quinoid pigment isolated from some sea-occurring invertebrates, which is used in ophthalmology and for the treatment of acute myocardial infarction and ischemic heart disorder. Seven new impurities were isolated from the bulk drug and its formulation and their structures were elucidated by NMR in combination with other spectroscopies. The physicochemical properties of the impurities are close to those of echinochrome. For this reason, they can be present in echinochrome bulk drug after technological operations of isolation and purification. Some of these are also natural products [89].
NMR spectroscopy was employed to identify and quantitate impurities in dihydroquercetin (DHQ) [90]. Quercetin, related flavonoids (naringenin, kaempferol, dihydrokaempferol) and other species, resulting from plant raw material extraction, and impurities resulting from the extraction process, such as acetone, could be identified and quantitated from the proton spectra.
The qualitative and quantitative composition of the impurities is unique for the raw material and the DHQ production method. Therefore, it was proposed that 1H NMR spectra are good fingerprints for determining the origin and preparation method of drugs based on DHQ.
Salidroside has been widely used in Chinese traditional medicine for its antioxidant and antiapoptotic properties and is a drug candidate for the treatment of cardiovascular and cerebrovascular diseases. Three impurities with related chemical structures were found during the analysis of the bulk drug and characterized by various 1D and 2D NMR techniques and other spectroscopies [91]. A pathway for their biosynthesis was also proposed, as shown in Scheme 1.
Scheme 1.
Biosynthetic pathway of salidroside and its impurities.
Sodium tanshinone IIA sulfonate (STS) is a water-soluble derivative of tanshinone IIA, an important lipophilic component isolated from the roots of Salvia miltiorrhiza. As an injection, STS is widely and successfully used in China for treating cardiovascular diseases. In the analysis of STS bulk drug, eight related impurities were observed and isolated by column chromatography, which structure was established employing a combined NMR and LC–ESI/MS approach [92]. NMR experiments (1H, 13C, HMBC, HSQC, COSY) were crucial for unequivocally establishing structural features, where MS was not useful, allowing to propose a possible mechanism for the formation of the impurities. Some of the impurities found were known related compounds [93].
NMR spectroscopy was also employed for the structural elucidation of previously unknown impurities coming from new isolation processes. The traditional procedures for isolating vincristine and vinblastine from Catharanthus roseus are relatively inefficient. Recent efforts have resulted in a more effective manufacturing process at the expense of the appearance of new impurities. These and other known impurities were unambiguously identified by NMR spectroscopy, combined with MS techniques[94] and [95], after preparative chromatographic isolation of impurities. An 800 MHz NMR spectrometer equipped with a 1H{13C/15N} Triple Resonance 13C Enhanced Salt Tolerant Cold Probe was employed, where 2D experiments could be run overnight with 10 mg samples. 1H–1H, direct 1H–13C, long-range1H–13C scalar and dipolar spin–spin connectivities were established from a combination of 1D (1H, 13C) and 2D (gHSQCAD, zTOCSY, gHMBCAD, NOESY and ROESY) NMR experiments. A DBPPSTE DOSY diffusion experiment was also performed.
Collection of the required spectral data was made more difficult by the relatively large size of the molecules, which yielded crowded spectra, turning laborious the extraction of spin–spin connectivities. In addition, these bisindoles exhibit solvent-, field-, and temperature-dependency of the sign and size of the 1H–1H NOEs, serious solvent-dependent line-broadening that affects different signals differently in any given solvent, or line splitting due to slow N-formyl rotamerism in vincristine analogs. These difficulties were overcome by running the experiments in two or three different solvents and using the whole set of data in a complementary fashion in order to extract all the required information.
Applying the mass spectrometric shift technique to HPLC–ESI–MS/MS analysis, four impurities were identified in the unique semi-synthetic vinca alkaloid vinorelbine bitartrate. These were isolated by prep-HPLC and their structures were further confirmed by means of 1D and 2D (HMBC, HMQC, COSY) NMR spectra [96].
Most amino acids lack proper chromophors for UV-detection; therefore, the determination of low levels of impurities is analytically challenging [97] and [98]. Amino acids are produced by different manufacturing processes, each one conveying to the product a different impurity profile, with fermentation resulting in the most complex array of impurities [99]. NMR was employed as an orthogonal technique to HPLC for quantitative analysis of potential impurities of alanine. Organic acids such as malic, fumaric, aspartic and glutamic, were assessed by 1H NMR spectroscopy with 400 and 600 MHz spectrometers using 128 and 16 scans, respectively [35] and [98]. In addition, L-Isoleucine and L-leucine present as impurities in L-valine are responsible for some of the process impurities found in lopinavir [100]. The latter were characterized by FT–IR, MS and 1H NMR techniques.
During the 2007–2008 heparin crisis it was detected the presence of oversulfated chondroitin sulfate (OSCS), the adulterant of the API which caused many deaths [101]. NMR spectroscopy has been in use for fractionated medium-sized heparins more than a decade before this case. This event, however, where NMR spectroscopy played a decisive role in the structural determination of the contaminant, spurred intense research activity in this area, which resulted in a series of tests orthogonally aiming to achieve identification, measurement of bioactivity and detection of process impurities or contaminants in these drug products [102]. As a result, the current enoxaparin USP monograph contains a 13C NMR spectroscopy test and the heparin sodium monograph includes a 1H NMR identification test [103].
1H NMR spectroscopy is very sensitive to minor structural variations, allowing easy identification of the repeating pattern of the disaccharide units of heparin. Therefore, not surprisingly NMR spectroscopy proved to be a powerful tool for quali- and quantitative analysis of compounds related to the extraction and purification processes of heparin, including dermatan sulfate, chondroitin sulfate A, galactosamine and 3-O-acetylated uronic acid derived impurities [104], [105], [106] and [107].
In these cases, major advantages of NMR analysis are its unique ability to unambiguously determine sulfation locations and the stereochemistry of the anomeric bonds between the saccharide subunits. The most useful experiments for spectral assignment are COSY and TOCSY (homonuclear correlation), NOESY and ROESY (nuclear Overhauser effect), HSQC and HMQC (heteronuclear single or multiple quantum coherence spectroscopy). Fortunately, some of the characteristic resonances of these contaminants lie outside the heparin regions, easing their detection; chemometrics analysis of the data resulted in more powerful methods [108], [109], [110] and [111].
Besides the use of NMR spectroscopy for impurity detection and characterization, scattered articles have reported the use of qHNMR as a way to determine the purity of isolated plant metabolites, along with the identity of their impurities [112], [113] and [114].
2.1.2. Impurities in synthetic active pharmaceutical ingredients
APIs are usually prepared through multistep organic synthesis. Process-related impurities can be formed at any step and can ultimately appear in the final bulk drug, especially during scale-up of early-stage drug candidates. These low-level impurities formed by side reactions are always problematic, since they may affect the API through an unwanted pharmacologic activity or just merely from the cosmetic point of view, when colored by-products are formed. Therefore, it is always desirable to investigate the structures of by-products not only in the reactants but also in intermediates and final products. Elucidation of the structures of these process impurities is an important step in refining the process chemistry in order to develop robust routes for manufacturing APIs.
Different techniques, including LC coupled to ion-trap and time of flight mass spectrometry (IT/MS, LC–TOF/MS) and NMR (1H, 13C, DEPT, and HETCOR) allowed characterization of impurities in chloroquine and hydroxy chloroquine bulk drug samples [115]. Process-related impurities were also detected in ezetimibe by LC–MS/MS and elucidated by NMR. NOESY experiments were relevant in this investigation[116] and [117]. An impurity was found in phenazopyridine hydrochloride; its identification by MS–MS and 2D-NMR spectroscopy allowed to propose a mechanism for its formation [118]. Analogously, a tridesoxy impurity found in didanosine and characterized by 300 MHz NMR (1H and 13C) was proposed to result from deoxygenation during the hydrogenation stage of the preparation of the API [119].
Six process-related impurities were detected in pantoprazole sodium bulk drug substance [120] and another six impurities were found in the structurally related rabeprazole [121]. These impurities differed mainly in the degree of oxidation of both, the sulfur bridge and the pyridinic nitrogen. They were identified, synthesized and characterized using 1H, 13C and DEPT NMR experiments, aided by techniques such as HPLC, LC–MS and IR. Two impurities of tazarotene were characterized by NMR [122].
Process-related impurities found in triclabendazole were identified by 1D and 2D NMR (1H, 13C and DEPT, HSQC, HMBC) spectroscopy, concluding that one of the impurities originates in an impurity of the starting materials [123]. Glutamine as an impurity of glutamic acid generated a process related impurity in leucovorin [124]. Synthesis and spectroscopic characterization (IR, MS, 1H and 13C NMR) of the impurity provided the definitive proof for its postulated structure. The process related impurities in ceftizoxime sodium bulk drug were identified, isolated and characterized by using HPLC (analytical and preparative), LC–MS–MS, IR and NMR (1H, 13C) techniques. Spectra of the impurities and the API were compared [125].
Process-related impurities were also found in telmisartan [126], valsartan [127] and candesartan cilexetil[128], and elucidated/confirmed based on 2D-NMR and MS data. Three impurities were detected in simvastatin substance and tablets By HPLC–DAD. Their proposed structures revealed modifications of the lactone ring of the simvastatin molecule; one of them was synthesized and structurally characterized by NMR [129]. For the development of an HPLC separation, process-related impurities of pridinol mesylate were also synthesized and characterized by 1H and 13C NMR spectroscopy [130].
Classical methods were also employed for the isolation and characterization of process related impurities in eslicarbazepine [131]. A potential process related impurity of phenazopyridine HCl was isolated by preparative HPLC and structurally elucidated by MS–MS and 2D-NMR spectroscopy [118]. Analogously, four process-related impurities were found in the novel antiepileptic drug retigabine [132], which included (Scheme 2) the reduced unreacted starting material (Impurity-1), a defluorinated analog (Impurity-2), the solvent-acylated final product (Impurity-3) and the last intermediate (Impurity-4). Their structures were determined by 1D (1H, 13C, DEPT, 19F), and 2D (1H–1H COSY, HMBC, HSQC) NMR experiments.
Scheme 2.
Synthetic sequence of the manufacturing process of retigabine and sources of the impurities.
Eight related impurities of olanzapine were isolated and characterized, including a new impurity confirmed as 1-(5-methyl-thiophen-2-yl)-1H-benzimidazol-2(3H)-one by X-ray single diffraction, MS, 1H NMR, 13C NMR and HSQC [133]; a mechanism for its formation was proposed.
The structural determination [IR, MS and NMR (1H, 13C, DEPT)] of four impurities in zafirlukast allowed to propose a synthetic scheme to account for their origin [134]; analogously, four impurities were found in montelukast; after their characterization by spectroscopic analysis and the corresponding synthesis, their origin was also proposed [135].
Some examples found in recent literature point out to special precautions required during complex syntheses. The structural analysis (MS, IR, and 200 MHz 1H and 13C NMR) of an impurity isolated from lisinopril revealed it to be a result from incomplete protection of the starting material [136]. An impurity was isolated from benazepril and subjected to spectroscopic analysis using LC–MS/MS, MS, NMR (1H and 13C) and FT-IR. From its structure, it was proposed that it might have been generated due to the presence of small amounts of moisture during a key hydrogenation step [137].
Zolmitriptan-dimer, which was identified as by-product of the last step Fischer indole synthesis, was characterized as an impurity in zolmitriptan by means of LC–MS and NMR studies [138]. Two new dimers (1,2 and 2,2) were found as process impurities in rizatriptan. From the 1H and 13C NMR values it was concluded that N,N-dimethyl butanal diethyl acetal which is used as reagent in the synthesis of rizatriptan is reacting at indole 2-position of one rizatriptan molecule and at reactive positions of another rizatriptan molecule, forming a dimer with loss of ethanol [139].
Three process impurities were observed in adapalene and characterized by 1H and 13C NMR spectroscopy. Taking into account the synthetic process of the API, one of them is formed during the Friedel–Crafts step employed to prepare the first intermediate, while the others are formed during the Negishi coupling of the latter. Their presence is diagnostic of the involvement of these steps in the corresponding synthetic procedure [140].
After a systematic revision of the synthetic process, the structural elucidation and quantitative determination of seven process impurities (two of them new) of the melatonergic agonist agomelatine was carried out, employing reference standards of the impurities and with the help of NMR and FT-IR techniques [141]. This allowed the development of a new chromatographic method for analysis of the API (Scheme 3).
Scheme 3.
Manufacturing process of agomelatine and origin of the potential impurities originating in side reaction and degradation.
Relevant impurities were found in citalopram and escitalopram; their structures were elucidated by MSn(ion trap mass analyzer), accurate mass (Q-TOF mass analyzer), FT-IR and NMR (1H, 13C NMR and DEPT) data [142] and [143]; this enabled to propose a mechanism for their formation. A process impurity in a crude roflumilast preparation was isolated, characterized by NMR spectroscopy and its identity confirmed by an independent synthesis [144].
Four process and degradation impurities were found in darifenacin. Based on the structures of the impurities, proposed by LC–MSn, these were independently synthesized and used for further structural confirmation by IR, NMR and MS techniques [145].
2.1.3. Process impurities: Geometric and conformational isomers
The geometric isomer Z of endoxifen was confirmed as the active isomer through 2D NMR experiments, including HSQC and COSY [146]. NOE (ROESY) experiments were employed to establish spatial proton–proton correlations between key protons on the ethyl group and the neighbor aromatic rings and confirm the stereochemistry of each isomer ( Fig. 1).
Fig. 1.
1H and 13C NMR diagnostic signals of Z– and E-endoxifen (ppm values).
The major impurity of the drug substance was found to be the E-isomer. The previous identification of the geometric isomers of endoxifen by 1H NMR was empirically based on reported trends for the chemical shifts of the OCH2 protons in the side chain, mentioned to be upfield in the (Z)- compared to (E)-configuration for similar triarylethylene compounds [147].
The identification and characterization of a geometrical isomeric photo degradation product of eprosartan was reported, using LC–MS and LC–NMR [148].
Five impurities were isolated from a crude reaction mixture of glimepiride. These were spectroscopically characterized by NMR, FT-IR, UV and MS; among them, the cis (geometric isomer), as well as the metaand ortho (positional) isomers were found [149]. On the other hand, the 1H NMR spectrum of enalaprilat exhibits two separate (Δδ ≈ 0.2 ppm) sets of signals. They correspond to the cis and trans conformers of the drug [150], which are in equilibrium with a isomer ratio of 71.5:28.5 at 298 °C, and arise from the slow rotation around the amide bond.
2.1.4. Process impurities: Positional and functional group isomers
A positional isomer of primaquine was detected as an impurity. The proposed compound was synthesized and characterized by NMR spectroscopy [151]. Analogously, four impurities were detected in piperaquine phosphate API by a newly developed gradient RP–HPLC method. They were identified by LC–MS/MS and their structures, which relate to the manufacturing process, were confirmed by NMR and FT-IR spectroscopy, conducted using synthesized authentic compounds. A positional isomeric impurity was identified by 2D NMR spectroscopy (1H–1H COSY, HSQC, HMBC). This impurity is formed because of contamination of batches of the 4,7-dichloroquinoline precursor with 4,5-dichloroquinoline [152] and [153].
LC–MS was employed to detect an isomeric impurity in cefpodoxime proxetil, a third generation cephalosporin, and its hydrolysis product. Detailed structural information was confirmed by LC–NMR [154]. Isomeric impurities in cefdinir were confirmed in an analogous fashion [155].
Ivermectin is produced by hydrogenation of a series of avermectins. Examination of ivermectin by LC–MS data revealed four process impurities. In one of them the fragmentation pattern was identical to avermectin B2a indicating a possible isomer; this suspicion was confirmed by NMR spectroscopy [156].
The related compounds of the novel hypoglycemic drug G004 were identified. Using 1H, 13C NMR and 2D NMR analysis and MS data, it was found that one of the isolated compounds was an isomer of the drug[157]. Etimicin sulfate is a semi-synthetic aminoglycoside prepared from gentamicin C1a. Ten impurities were detected in the bulk drug by LC–ELSD and LC–ESI–MSn, many of them followed the similar fragmentation pattern as a result of differing in the position of an ethyl substituent. Six of them were isolated by column chromatography and two impurities proved to be isomers of the drug after 1H and 13C NMR analysis and comparison with literature data [158].
1H and 13C NMR spectral analysis helped elucidating a C-alkylated isomeric impurity (functional group isomer) in duloxetine hydrochloride [159] and [160], a O-alkylated naphthol ( Fig. 2). The formation of this unusual reaction product was attributed to the ambidentate nature of the nucleophile naphthol.
Fig. 2.
Chemical structures and diagnostic NMR resonances (in ppm) in duloxetine and its impurity.
2.1.5. Process impurities: Configurational isomers. Epimers and enantiomers; NMR determination of enantiomeric purity
Molecules having stereogenic centers are highly important as pharmaceutical products; some of them are natural products, but semi-synthetic and fully synthetic small organic molecules constitute an increasing proportion of these APIs in the current pharmaceutical arsenal, many of which are sold as a sole optically active enantiomer. The ICH Q6A guideline states that “where a new drug substance is predominantly one enantiomer, the opposite enantiomer is excluded from the qualification and identification thresholds given in the ICH Guidelines on Impurities in New Drug Substances and Impurities in New Drug Products because of practical difficulties in quantifying it at those levels. However, that impurity in the chiral new drug substance and the resulting new drug product(s) should otherwise be treated according to the principles established in those Guidelines” [161].
Accordingly, the quality control of chiral reagents and products requires methods capable of discriminating between enantiomers and for determining their enantiomeric purity, to resolve questions such as whether racemization has occurred during the synthesis of a chiral molecule or distinguish the enantiomeric impurity among other impurities in a sample. Typically, a 98% pure catalyst, ligand or auxiliary reagent will intrinsically afford at least 2% of the wrong enantiomer, lowering the enantiomeric excess of the final product. Therefore, since 1998 the control of impurities in chiral reagents [162], [163] and [164], involves NMR among other methodologies, is a routine task.
Currently, the method of choice for limiting enantiomeric impurities is chromatography (GC, HPLC) with chiral columns. Among the other alternatives, NMR spectroscopy is the only non-separative technique useful for the determination of the stereochemical purity of materials of pharmaceutical interest, employing for that purpose chiral derivatizing agents, lanthanide shift reagents and chiral solvating agents (CSA). CSA-based chiral NMR methods can be developed when only a single enantiomer of the analyte is available [165], provided both enantiomers of the CSA are at hand. This and other developments have converted 1H NMR into a straightforward and more time-efficient approach toward detection and quantification of enantiomers [166] and [167].
NMR spectroscopy is also well known to be one of the most powerful tools for the elucidation of the chiral recognition mechanism on a molecular level [168]. NMR signal separation in chiral environments has been studied and the corresponding mechanisms were related to those operating in separative techniques such as HPLC and CE [169]. The chiral recognition can also be observed by 13C NMR spectroscopy [170]; however, since this is a less sensitive alternative, the method is not used as frequently as 1H NMR.
The enantiomeric purity of acyl-l-carnitine was determined by 500 MHz 1H NMR employing fast diastereomeric interaction with chiral shift reagents such as chiral lanthanide-camphorato {[Eu(hfc)3], [Pr(hfc)3]} or chiral samarium-pdta shift reagents. The LOD of the determination was 0.5% for the D-enantiomer [171].
NMR is also a powerful tool for distinguish between diastereomers. Seven related impurities were detected in asperosaponin VI bulk drug and their structures were elucidated by TOF-MS, IR and NMR techniques. One of the impurities was found to be a mixture of two epimers (α- and β-glucosyl anomer derivatives)[172]. Four impurities were detected in the semisynthetic drug docetaxel. Two of them are oxazolidinones resulting from side reactions during the introduction of a Boc protecting group and acid deprotection, while the remaining pair are epimers of the API, formed by isomerization of two different hydroxyl groups[173] and [174].
Three process impurities were recently detected in the fluorine-containing steroid clocortolone pivalate by HPLC. They were isolated by semi-preparative LC and identified by MS and NMR spectroscopy. One of them was the 6β-fluor epimer of the API. Their formation was also proposed [175]. Table 2 summarizes the role of NMR spectroscopy in providing improved knowledge on process impurities of pharmaceutically relevant active ingredients.
Table 2.Process impurities found in some active pharmaceutical ingredients.
| Drug | Field (MHz) | NMR experiments | Remarks | Ref. |
| Agomelatine | 500 | 1H, 13C, DEPT, COSY, HMBC, HSQC | Imp-1: First intermediate.Imp-2 and Imp-3: Side reaction products of Imp-1.Imp-4: Acetylated derivative of Imp-2.Imp-5: Starting material.Imp-6: Dimer of Imp-1.Imp-7: Acetylated derivative of Imp-6. | [141] |
| Chloroquine (CQ) Hydroxychloroquine (HCQ) | 400 | 1H, 13C, DEPT, COSY, HETCOR (1H–13C) | Imp-1 and Imp-2 (CQ-I and HCQ-I): Formed by deethylation of intermediates 5-(N,N-diethyl-ethylamino)-2-pentyl amine and 5-(N-ethyl-N-2-hydroxyethylamino)-2-pentyl amine, respectively.Imp-3 and Imp-4 (CQ-II and HCQ-II): Quaternary ammonium products formed by chloromethylation of the amino side chain of the APIs in basic medium, with dichloromethane. | [115] |
| Citalopram | 400 | 1H, 13C, DEPT-135 | Imp-1: The Grignard reagent performs a double addition in the first step of the synthesis. The product undergoes all the following reactions toward the API.Identified as 1-(1,1-bis(4-fluorophenyl)-1,3-dihydro isobenzofuran-5-yl)-4-(dimethylamino) butan-1-one HBr. | [142] |
| Clindamycin palmitate hydrochloride | 300 | 1H, 13C, DEPT | Imp-1 (Clindamycin palmitate sulphoxides α/β-isomers): Formed by oxidation at the sulfur atom.Imp-2, Imp-4, Imp-7 Imp-9 and Imp-10 (laurate, myristate, pentadecanoate, heptadecanoate and stearate ester of Clindamicin respectively): From impurities in the starting palmitoyl chloride.Imp-3 (Lincomycin palmitate): Lincomycin is the starting material for the synthesis of Clindamycin.Imp-5 (7-epiclindamycin palmitate): Formed by C-7 epimerization.Imp-6 (Clindamycin palmitate 3-isomer): Formed by esterification on C-3.Imp-8 (Clindamycin B-palmitate): Clindamycin B present as an impurity in the starting Clindamycin.Imp-11 and Imp-12: Starting palmitic acid and Clindamycin. | [177] |
| Clocortolone pivalate | 200, 300, 600 | 1H, 13C, NOESY | Imp-1: 6β-Fluor epimer.Imp-2: 4-Fluor isomer.Imp-3: 11-Pivaloylated Δ60,07 derivative. | [176] |
| Escitalopram | 400 | 1H, 13C, DEPT, HMBC, HSQC, DQF-COSY | Imp-1 (ESC-I): Uncyclized precursor of the API.Imp-2 (ESC-II): Formed by chloromethylation of the amino side chain of ESC with CH2Cl2(solvent).Imp-3 (ESC-III): Formed by chloromethylation of the amino side chain of ESC-I with CH2Cl2. | [143] |
| Eslicarbazepine acetate | 400 | 1H, 13C, COSY | Imp-1: Due to the presence of propionic anhydride in acetic anhydride, used in the last step of the synthesis. | [131] |
| Ezetimibe | 500 | 1H, 13C, DEPT, COSY, HSQC, HMBC | Imp-1: Formed during hydrogenation of the last intermediate (benzyl ezetimibe), by deoxygenation of the benzyl alcohol with cleavage of the β-lactam C-N bond.Imp-2: Formed by deoxygenation of the benzyl alcohol moiety. | [117] |
| G004 | 500 | 1H, 13C, HMBC | Imp-1: 1-(4-(2-(2-bromobenzenesulphonamino) ethyl) phenylsuphonyl)-3-(trans-4-methyl cyclohexyl) urea is the 2-bromo analog. Isolated using LC. | [157] |
| Montelukast sodium | 500 | 1H, 13C, DEPT, 2D (unspecified experiments) | Imp-1: Result of incomplete transformation of the cyano group into a carboxate during the basic hydrolysis stage.Imp-2: Formed during the Montelukast work up stage by protonation and subsequent dehydration of the tertiary hydroxyl moiety.Imp-3 and Imp-4: Saturated and deschloro analog of the starting material, which undergo all the sequence of reactions leading to the API. | [135] |
| Naproxen | 400 | 1H, DQFCOSY, gHSQC, gHMBC | Imp-1: Identified as 2-(6-methoxynaphthalen-2-yl) acrylic acid, the acrylic acid of naproxen. | [33] |
| Phenazopyridine.HCl | 400 | 1H, COSY, NOESY | Imp-1: Formed by addition of phenyl radical to phenazopyridine HCl during last step of the synthesis. Identified as 3-phenyl-5-phenylazo-pyridine-2,6-diamine. | [118] |
| Retigabine | 400 | 1H, 13C, 19F, COSY, HMBC, HSQC | Imp-1: Produced by reduction of ethyl 2-nitro-4-aminophenylcarbamate as a vestigial intermediate.Imp-2: Resulting from benzaldehyde being an impurity in 4-fluoro benzaldehyde.Imp-3: Acetylation product of retigabine.Imp-4: Vestigial intermediate of retigabine. | [132] |
| Rizatriptan Benzoate | 300 | 1H, 13C | Imp-1: Formed during the Fischer indole reaction, rizatriptan further undergoes a dimerization reaction through electrophilic substitution, yielding 1,2 and 2,2 rizatriptan dimers. | [139] |
| SodiumTanshinone IIA Sulfonate | 300, 500 | 1H, 13C, COSY, HMQC, HMBC | Imp-1 and Imp-5: Hydroxylated derivatives of the API.Imp-2: Formed by removal of the methyl group at C-4 of Imp-4.Imp-3 and Imp-6: Side products attributed to oxidation of C-19.Imp-4: Formed by dehydration of Imp-1.Imp-7: Formed by hydration of the furan ring of Imp-1.Imp-8: Produced by oxidation and further esterification at C-19 of Imp-4. | [92] |
| Tazarotene | 300 | 1H, 13C | Imp-5: Side reaction of a PCl3-mediated Pummerer rearrangement.Imp-6: Side product of a Sonogashira coupling reaction. | [122] |
| Telmisartan | 300 | 1H, 13C, DEPT, COSY, HSQC, HMBC | Imp-1: Formed during radical bromination of the starting material 4-methyl biphenyl-2-carboxylate. | [126] |
| Valsartan | 400 | 1H, 13C, DEPT | Imp-1 and Imp-2: Synthetic intermediates.Imp-3 and Imp-4: Products resulting from free radical desulfurization.Imp-5: Result from reaction of 5-phenylthiovaleric acid with Imp-1 and subsequent alkaline hydrolysis. | [127] |
| Vincristine (VCR)Vinblastine (VLB) | 800 | 1H, 13C, 15N, COSY, HSQC, NOESY, ROESY, HMBC, gHMBCAD, 1H–15N gHSQCAD, zTOCSY | Three new impurities.Imp-2: Cyclo-VCR.Imp-4: [VCR]-C(16)-COOEt, from oxidization of Imp-5.[VLB]-C(16)-COOEt: This impurity is a new natural product. | [94] and [95] |
| Zafirlukast (ZLK) | 200, 400 | 1H, 13C, DEPT | Imp-1: Formed by trans-esterification of the cyclopentyl group of ZLK with methanol under basic conditions.Imp-2: Formed from Imp-1 by condensation with o-toluene sulfonamide.Imp-3 and Imp-4: Due to the presence of m/p-toluene sulfonamide impurities in o-toluene sulfonamide.Imp-5: Synthetic intermediate. | [134] |
2.1.6. Genotoxic, toxic and mutagenic impurities
Genotoxic impurities (GTIs) are chemical compounds that may be mutagenic and could potentially damage DNA with an accompanying risk of cancer.
The analysis of GTIs has many special characteristics and difficulties. First of all, it can be very challenging because usually they must be controlled at levels significantly lower than 0.1% (1000 ppm). According to the current guidance, the threshold for toxicological concern in commercial products is 1.5 μg/day. Current regulations expect to control GTIs to levels in the ppm range [176]. Therefore, the analytical procedure should ideally allow detection limits in the range of 1–5 ppm.
Another special issue is solubility, since high sample concentrations are required (100 mg/ml) to enable analyses of the low-level GTIs. This poses the risk of sample precipitation, especially if the sample solution needs to be cooled in order to control degradation.
In addition, there is no single analytical procedure for analysis of all GTIs, because of their wide structural diversity. Furthermore, their reactive and often volatile nature poses difficulties on sample preparation in order to ensure the integrity of the analytes.
Therefore, the analyst should always decide on which is the correct analytical technique for each particular GTI determination. Among the five major alternatives (GC, LC, GC–MS, LC–MS and NMR), the GC-based techniques are preferred for the most volatile compounds, and the MS-based alternatives should be used for the cases requiring the highest sensitivity.
The chromatographic techniques are very complementary with each other. However, NMR has areas of application which can overlap with all of them, being most suitable when the compounds are not too volatile and their abundance is not too low [178].
The advantages, disadvantages and potentials for the use of NMR in GTI analysis have been recently discussed in depth [179]. Therefore, with the modern digitizers, the LOD and LOQ in qNMR are dependent on the number of scans, the solubility of the API and the strength of the magnetic field.
Although not many examples are currently available to illustrate the potential of NMR spectroscopy in this area, it is clear that this is one of the methods that will increase in importance in the coming years for detecting and quantitating these impurities.
A potential genotoxic impurity from the synthesis of an API was used to challenge the LOD/LOQ on a 400 MHz spectrometer with a limit test. Samples were spiked with the impurity up to 100 ppm and spectra were acquired (4096 scans, D1 = 2 s) with the inverse-gated 13C decoupling sequence to prevent overlapping satellites with the impurity. Unequivocal detection of the spiked genotoxic impurity was demonstrated at a level of 25 ppm [180]. In addition, qNMR was used for quantitation of methanosulfonic acid throughout the production process of an API and a practical quantification limit of 100 ppm was consistently attained [181].
Hydrazines, hydrazides and hydrazones must also be controlled in APIs [182]. Aryl hydrazone isomerization, resulting in the formation of residual geometrical E-isomer impurities, were determined by HPLC with DAD and MS–MS detection, followed by confirmatory NMR data [183].
Combined application of LC and NMR spectroscopy allowed characterization of mutagenic impurities in vestipitant. The starting material for the synthesis was identified as the root cause of impurities [184].
Finally, a toxic impurity was isolated from the experimental anti-neoplastic drug XP315. Characterization by combination of NMR (400 MHz, including gCOSY, gHMBC, gHSQC) and mass spectral methods revealed that it resulted from the dimerization of an intermediate by ring fusion, during the synthetic process [185].
2.1.7. Organic volatile impurities (residual solvents) and other accompanying impurities
The organic volatile impurities (OVIs) are a potential toxic risk for pharmaceutical products and have been a concern of manufacturers for many years. The ICH addresses this topic in its Q3C guideline [186]. The most widely used method for analysis of OVIs is GC [187]; however, there has been a slow but increasing use of NMR in this area. When the first coupled continuous-flow capillary GC–solenoidal microcoil 1H NMR spectroscopy (400 MHz) experiments were disclosed, the separation of a mixture of diethyl ether, dichloromethane and tetrahydrofuran, at an oven temperature of 60 °C was informed [69]. Acetone, resulting from the extraction process was identified and quantitated by NMR spectroscopy in dihydroquercetin, isolated from natural sources [90].
Preparation by-products such as ethanol and acetone are among the most common impurities identified in heparin preparations by 1H and 13C NMR spectroscopy [104], [188] and [189]. Being a process impurity, for the development of a chemometric-NMR approach to heparin analysis, the presence of EtOH was considered a potential interference for the determination of the drug or other impurities [106]. Other residual solvents like methanol, formic acid [104], and butanol (δ = 0.91, 1.35, 1.53, and 3.61 ppm) were also found in heparin samples [190], as well as sodium acetate and the chelating agent ethylenediaminetetraacetic acid (EDTA) (δ ∼ 3.5 ppm) [191].
It has been reported [192] that when initial studies concluded that direct injection or headspace GC could not be employed to determine the levels of N-methyl pyrrolidinone (NMP) in a process intermediate, a 1H NMR assay was developed and validated to directly quantitate the solvent at a 140 ppm level in the sample matrix.
Aldehydes and silicon derivatives have also been observed by NMR spectroscopy. Bortezomib is a modified dipeptidyl boronic acid [193]. Recent comparison between two brands of the drug revealed the presence of tert-butanol, a compounding solvent. In addition, this API proved to have silicon-containing impurities such as silanes and siloxanes and isovaleraldehyde as an impurity of unknown origin [194].
The related aldehydes, acetaldehyde and propionaldehyde, were quantitatively determined as impurities in poloxamer 188 by 1H NMR [195]. On the other hand, the NMR detection limit of residual silicone oil in poly(lactic acid) and poly(lactic acid–co-glycolic acid) microspheres, was determined to be 100 ppm, which compared favorably with IR determinations (5000 ppm) [196].
NMR spectroscopy was employed to determine the levels of residual benzene, toluene, acetone, and ethyl ether in cocaine [197]; ethyl acetate and methylene chloride, which were previously not detected in cocaine samples, were also found. An excellent correlation was obtained between the results of Schöniger flask combustion analysis for chlorine as an appraisal of chlorinated solvent levels and NMR-based estimates of chloroform. This demonstrated the suitability of NMR spectroscopy for the quantitation of chlorinated solvents [198].
2.2. Degradation products
2.2.1. Impurities resulting from degradation of synthetic APIs
Forced degradation is a useful tool for impurity profiling [199] and [200]. Due to its multiple advantages, NMR spectroscopy is widely used in pharmaceutical analysis in order to understand the course of the degradation and also identify and structurally elucidate its embedded impurities.
Stress tests of eprinomectin, a synthetic acetylated amino derivative of abamectin, revealed two impurities. Given their isomeric nature (2-epimer and Δ2,3-isomer) their structures became fully elucidated only after analysis of 1H, 13C, NOESY and COSY experiments [201].
The novel antiepileptic drug carisbamate undergoes significant degradation when subjected to stress conditions. The degradation products were isolated and characterized. MS/MS and 2D-NMR (COSY and HSQC) studies revealed that one of the degradation products resulted from positional isomerization of the carbamate moiety of the API [202], being prepared as shown in Scheme 4.
Scheme 4.
Synthesis of carisbamate and origin of its isomeric impurity.
Twelve impurities of clindamycin palmitate hydrochloride were isolated by preparative HPLC and characterized by a combination of LC–MS, FT-IR and simple 1D NMR (1H, 13C and DEPT) techniques[176]. Thorough NMR analysis was used for the characterization of an oxidation impurity of clopidogrel[203] and impurities of pridinol mesylate [204].
An impurity of citalopram was isolated by semi-preparative HPLC and its structure was established by MS, NMR and IR spectroscopy [142]. Analogously, chloroquine-N-oxide was identified as the major oxidative degradation product of chloroquine [205]. An N-oxidation product of cetirizine was isolated and characterized by 400 MHz NMR spectroscopy along with LC–MS/MS. The product is also a metabolite of the drug [206].
Two isomeric monoacylated diacerein impurities (MW = 326) were characterized by NMR spectroscopy, which confirmed their structures and revealed the exact position of the acetyl group in each case [207]. Unknown impurities of the antidepressant drug escitalopram were elucidated by 1H and 13C NMR and their structures were confirmed by synthesis [143]. Preparative HPLC, LC–MS/MS, UPLC–TOF-MS, NMR and FT-IR spectroscopy were employed to study the forced degradation of dipyridamole product, resulting in two new impurities (one of them an interaction product) which were characterized by NMR spectroscopy[208]. LC–NMR and LC–MS allowed to elucidate a new impurity in the antifungal drug icofungipen [209].
A novel impurity in eprosartan bulk drug was characterized by ESI/MSn and NMR [210], and five impurities were found in candesartan cilexetil subjected to stability and forced degradation studies. They were structurally elucidated employing NMR techniques [211]. Low-level unknown impurities in GW876008, a novel corticotrophin-release factor 1 antagonist were isolated and characterized employing LC–NMR and HR-NMR in conjunction with mass spectrometric experiments [212]. Similarly, degradation products of anastrozole [213] and etimicin sulfate were determined by LC–MS/MS and NMR spectroscopy and LC–ESI–MSn and NMR, respectively [158]. LC–NMR was employed for structural characterization of the oxidative degradation products of SCH 56592, an antifungal agent [214].
Two unknown impurities of moxidectin were isolated by flash chromatography during stress testing of the drug, and fully characterized by 1D (1H, 13C) and 2D (NOESY, COSY, HMBC and HSQC) NMR experiments [215].
The photodegradation of fleroxacin was investigated in different injections and solutions. The three major photodegradation products were detected and isolated by HPLC, and elucidated employing FT-IR, MSnand TOF-MS, as well as 1H, 13C, DEPT, and 2D NMR. Significant differences in the photodegradation process and photostability between the concentrated injection and the dilute injection have been demonstrated, and photo-degradation pathways were proposed [216].
Analogously, photo-irradiation of tablets containing pramipexole afforded pyrrolidine carboxylic acids (Impurities 1 and 2) and catechol (Impurity-6). The drug was also smoothly degraded in the methanolic solution yielding carboxylic esters; a possible degradation mechanism was proposed in which the reaction starts by a [2+2] cycloaddition of 1O2 to the thiazole ring (Scheme 5). Cleavage of the CC bond of the thiazole, followed by attack of the secondary amine attack to the carbonyl group affords a pyrrolidinic structure, which is solvolyzed to furnish carboxylic acids and methyl esters depending on the reaction medium [217].
Scheme 5.
Proposed photo-degradation mechanism of pramipexole.
A photolytic degradation product of telmisartan and degradation products of irbesartan were detected and characterized using LC–MS/TOF, LC–MSn, LC–NMR and on-line H/D exchange mass studies[218] and [219].
The structure of the major degradation product of ezetimibe was initially proposed by NMR spectroscopy (1H and 13C) and MS studies [220]. Shortly after, and without providing a structure, it was pointed out that the previously proposed structure was inconsistent with the reported spectroscopic data [221]. Finally, a third group of scientists [222] effected detailed analysis of 1H, 13C, COSY, HSQC, HMBC and NOESY experiments, proposing a structure which agreed with a previously reported compound; 19F NMR was also employed in this research [223] and [224].
The thermal and alkaline degradation products of meropenem were identified by NMR and MS analysis. The in vitro cytotoxicity assay against mononuclear cells demonstrated their cytotoxicity, rising safety concerns and suggesting that special care must be taken to avoid exposure of the drug to the degradation conditions during the handling and storage of the pharmaceutical preparation [225].
Table 3 summarizes the degradation and process impurities found in some active pharmaceutical ingredients, while Table 4 contains a brief list of degradation products found by submission of different pharmaceutical ingredients to stress or forcing degradation conditions.
Table 3.Degradation and process impurities detected in some active pharmaceutical ingredients.
| Drug | Field (MHz) | NMR Experiments | Remarks | Ref. |
| Asperosaponin VI | 500 | 1H, 13C, HSQC, HMBC, COSY, ROESY, TOCSY | Imp-1, Imp-2, Imp-5 and Imp-6: Natural impurities.Imp-4: Resulting from acid hydrolysis. Formed by loss of a unit of arabinose.Imp-7: Resulting from acid and basic hydrolysis. Formed by loss of two units of glucose.Imp-3: Mixture of two epimers that are isomers of Asperosaponin VI. | [172] |
| Bicalutamide | 200 | 1H | Imp-1 (I) and Imp-3 (III): Formed by basic degradation.Imp-2 (II) and Imp-4 (IV): Side products.Imp-5 (V): Starting material.Imp-6 (VI) and Imp-7 (VIII): Process intermediates. | [226] |
| Carisbamate | 300 | 1H, 13C, COSY, HSQC | Imp-1 – Imp-4: Process intermediates.Imp-5 and Imp-6: Degradation products. | [202] |
| Doxofylline | 300 | 1H, 13C | Imp-1: Starting material.Imp-2–6: Process intermediates.Imp-7 (DP-I): Resulting from acid degradation.Imp-8 (DP-II): Produced by basic and acid degradation. | [227] |
| Lopinavir | 300 | 1H | Imp-1 (RS1): Process intermediate.Imp-2 (RS2): Starting material.Imp-3 (RS3): Resulting from acid hydrolysis.Imp-4 (RS4) and Imp-5 (RS5): Side products and thermal degradation products, formed on extended storage at higher temperature.Imp-6 (RS6) and Imp-8(RS8): Impurities derived from the starting material.Imp-9 (RS9) and Imp-10 (RS10): Side products. | [100] |
Table 4.Degradation products observed after exposing APIs or their products to stress conditions.
| Drug | Field (MHz) | Experiments | Remarks | Ref. |
| Anastrozole | 300 | 1H, 13C | Imp-1: Formed by basic hydrolysis of both nitrile groups of the API (diacid).Imp-2: Formed by basic hydrolysis of one nitrile group of the API (monoacid). | [213] |
| Artesunate-Amodiaquine | 500 | 1H, 13C, COSY, HSQC, HMBC, TOCSY, NOESY | Imp-1: Rearranged derivative of anhydro dihydroartemisinin. | [229] |
| Biapenem | 500 | 1H, 13C, COSY, HSQC, HMBC | Imp-1 – Imp-3: Products of hydrolysis.Imp-2: Dimer AImp-3: Dimer B | [230] |
| Chloroquine | 500 | 1H, 13C, HSQC | Imp-1 (Chloroquine N-oxide): Resulting from oxidative degradation. | [205] |
| Clopidogrel | 400 | 1H, 13C, DQF-COSY, HSQC | Imp-1: Oxidation impurity.Imp-2: Clopidogrel related compound A.Imp-3 (Related compound B1) and Imp-4 (Related compound B2): Positional stereoisomers of the API.Imp-5 (Related compound C): Optical isomer. | [203] |
| Dipyridamole | 400 | 1H, gDQCOSY, gHSQC, gHMBC | Imp-1 (DPI): Formed by hydrolytic loss of a piperidine moiety resulting under acidic and oxidative (peroxide) conditions.Imp-2 (DPII): Interaction product with tartaric acid formed during stress treatment under acidic, oxidative (peroxide) and thermal degradation conditions. | [208] |
| Eprinomectin (EPM) | 400 | 1H, 13C, NOESY, COSY | Imp-1 (Unknown 1): Alkaline degradation product. Identified as the 2-epimer of EPM.Imp-2 (Unknown 2): Alkaline degradation product of Imp-2. Identified as Δ20,03-EPM. | [202] |
| Estradiol | 500 | 1H, 13C | Imp-1: Oxidative product of estradiol. | [231] |
| Ezetimibe | 400, 500 | 1H, 13C, gCOSY, gHSQC, gHMBC, 1D NOESY | The impurity (2R,3R,6S)-N,6-bis(4-fluorophe nyl)-2-(4-hydroxy phenyl)-3,4,5,6-tetrahydro-2H-pyran-3-carboxamide was mis-identified in previous publications. | [222] |
| Irbesartan | 500 | LC–NMR was used.1H, COSY(with solvent suppression) | Imp-1 (DP-I): Produced by acid degradation.Imp-2 (DP-II): Basic degradation product.Imp-3 (DP-III): Resulting from photo-acid degradation. | [219] |
| Meropenem | 500 | 1H, 13C, COSY, HSQC | Imp-1 (DP1): Thermal degradation product.Imp-2 (DP2): Alkaline degradation product. | [227] |
| Olmesartan medoxomil | 600 | 1H, 13C, using a 1H–13C/15N triple resonance inverse probe in stopped-flow LC–NMR. | Imp-1 (DP-1): Esterified dimer of Olmesartan. | [232] |
| Pramipexole hydrochloride hydrate | 400 | 1H, 13C, 2D (unspecified experiments) | Imp-1: Photo-degradation product. | [217] |
| Pridinol mesylate | 300 | 1H, 13C | Imp-1 (ELI): Dehydration product formed under acidic conditions.Imp-2 (NOX): Formed under oxidative degradation conditions. | [204] |
| Thiocolchicoside | 300 | 1H, 13C | Imp-1 (D1SO) and Imp-2 (D1SO2): Formed by oxidative degradation with m-CPBA.Imp-3 – Imp-5 (D2, D3, D4): Acid degradation products. | [233] |
2.2.2. Impurities resulting from degradation of natural product APIs
Artesunate is the hemisuccinic ester of dihydroartemisinin, a product resulting from the reduction of artemisinin. When held under ICH zone IV storage conditions during 6 months (40 °C and 75% RH), artesunate is known to yield several products above the threshold of identification [228].
Artesunate-amodiaquine bilayer tablets were subjected to heat treatment (70 °C, 14–30 days, 75% RH), furnishing a new and degradation product (Impurity B). This was purified by flash chromatography followed by semipreparative HPLC and unequivocally identified by 500 MHz NMR experiments (especially COSY, TOCSY and HMBC) as the tetrahydrofuranyl acetate-rearranged derivative of anhydrodihydroartemisinin (Fig. 3); MS analysis completed the identification.
Fig. 3.
Degradation of artesunate and three-bond H → C correlations observed in the HMBC spectrum of Impurity B.
Further, an authentic standard of the impurity was synthesized. An analogous rearrangement has been described to occur for artemisinin congeners during a biotransformation process [234]. The product has been shown to be a surrogate secondary marker for the Fe(II)-catalyzed radical reaction to rearranged tetrahydrofuranyl acetate products [235] and [236].
An isomeric 2-deoxy-4α-hydroxy-anhydrodihydroartemisinin derivative was also isolated. This product, a result of intrinsic thermal unstability of artesunate itself, is produced by the homolytic radical opening of the endoperoxide bond, a transformation already recorded for other artemisinin derivatives [228].
Tacrolimus (FK506) is an immunosuppressive drug used to avoid organ rejection in patients that have undergone organ transplantation. This is a potent drug which has a narrow therapeutic index. Following clinical reports suggesting that use of generic brands resulted in a significant reduction in the tacrolimus concentration/dose ratio in the plasma of liver and kidney recipients, a group of analytical methods were employed to assess the level of impurities and potency of products found in the US marketplace [237]. Ascomycin has immunosuppressant properties similar to tacrolimus, which fundaments its determination. In addition, it has been reported that the drug tautomerizes, yielding related rearrangement products[238] and [239], and the analysis can be further complicated by the presence of mixtures of interconverting rotamers [240].
After preparing standards of tacrolimus, ascomycin and the excipients commonly found in the pharmaceutical formulations and performing the 1H NMR spectral comparisons, no ascomycin was detected above the LOD (0.7 w/w%) nor other known impurities were observed in any of the products. This clearly indicated that the clinically observed difference does not originate from quality (purity and potency) differences between the generic and innovator tacrolimus.
Even established APIs still hide secrets that await to be uncovered. The structure of a labile estradiol-related degradant, isolated from a pharmaceutical formulation containing estradiol and norethisterone acetate was elucidated by off-line and on-line NMR [241] as well as MS techniques. The product, which proved to be unstable, contains a 9,10-epoxy function. Its secondary and tertiary decomposition products were also identified, and a new oxidative degradation mechanism was proposed, which provides an improved explanation for the formation of some oxidative secosteroid degradation products [231]. The study also cleared some literature ambiguities.
Erythromycin is a complex macrolide antibiotic mainly composed of erythromycin A and also of erythromycins B-F. Several related substances may be formed by fermentation along with the aforementioned compounds. Unknown compounds found in an erythromycin formulation were investigated by LC coupled to MS and NMR. The degradation products were structurally elucidated, especially employing an array of off-line NMR techniques (1D 1H and 13C, and 2D TOCSY, NOESY, COSY, HSQC and HMBC); two of the degradation products were new [242].
2.3. Miscellaneous impurities
2.3.1. Impurities and degradation products related to excipients
Examples are scarce, and include degradation products of the excipients themselves and a case where an excipient promoted degradation of an API. Interaction products involving excipients are detailed separately. An unknown degradation product found in non-MS compatible HPLC analysis of a drug formulation was studied using a multidisciplinary approach. NMR analysis revealed it to be 5-hydroxymethyl furfural, resulting from degradation of lactose [243].
In addition, 1H NMR approaches to the determination of free fatty acids [244] and [245] and peroxidation products [246] in pharmaceutical lipids were recently reported. The first method was demonstrated in different fatty oils, fat samples, waxes and oleyloleat. The lipids are a group of widely used pharmaceutical excipients, many of which also have therapeutic properties [247] and [248].
A constituent of the artificial flavoring of a losartan potassium extemporaneous suspension formulation was held responsible for the photosensitized degradation of the API in the drug product [249]. The structures of the degradation products were determined using a combination of preparative HPLC, LC–MS and NMR (1H, 13C and 2D) experiments.
2.3.2. Leaching impurities
Low levels of extractables have been observed in vials of interferon-α2b finished product, following periods of filling line down time. Their structures were elucidated as chlorinated biphenyl derivatives by MS and NMR spectroscopic methods and their origin was found in the Rehau RAU-SIK silicone tubing used on the Bosch filling line [250]. The structures of these impurities revealed that they are formed during the polymerization process used to make the raw material for the tubes.
2.3.3. Interaction products
During a stability study of an enema formulation containing 5-aminosalycylic acid (5-ASA) and citric acid, the formation of three new peaks was observed by HPLC–MS and further investigated by HPLC–SPE–NMR. These proved to be citric acid amides of 5-ASA; therefore, a recommendation was issued for avoiding the use of citric acid in liquid 5-ASA formulations [251]. Impurities in 5-ASA have also been elucidated by LC–NMR [252].
Analogously, one of the impurities found after submission of a dipyridamole drug product preparation to stress conditions was a tartrate ester, resulting from esterification with tartaric acid, employed as an excipient [208].
Different pharmaceutical preparations against the common cold containing phenylephrine (PHE) and saccharose were studied and new tetrahydroisoquinoline-type impurities were discovered, arising from a phenolic cyclization between the API with sugar-derived formaldehyde (Scheme 6). These were characterized by LC–MS and NMR techniques [253].
Scheme 6.
Formation of interaction products between phenylephrine and sugar-derived formaldehyde.
An amide interaction product between phenylephrine and maleic acid in a combined cold formulation containing phenylephrine hydrochloride and chlorpheniramine maleate, has been reported NMR analysis confirmed the structure of the correct isomer formed, the α-hydroxyamide [254].
In addition, a hydroxylamine-type degradation compound of carvedilol drug product was characterized by NMR; its generation was attributed to an interaction of the drug with polyvinyl pyrrolidone (PVP) in the presence of moisture [255].
3. Other NMR active nuclei
Fluorine (19F) NMR spectra are invaluable in the analysis of fluorine containing compounds [256].When the impurity profile of 16 commercial formulations of ciprofloxacin was studied, and characterized using 19F and 1H NMR spectroscopy, it was found that four to twelve fluorinated impurities, among them fluoride ion and two already known compounds, were detected and quantified in the formulations analyzed by 19F NMR [257].
The 19F NMR analysis of five pharmaceutical preparations of fluoxetine and fluvoxamine revealed that one of the samples contained an unidentified fluorinated impurity [258]. NMR spectroscopy (1H, 13C, 19F,1H–1H, 1H–13C, HMBC) and especially a nOe experiment helped to elucidate the place where substitution of a fluorine by a dimethylamino group took place in fluconazole (Scheme 7), resulting in a new, monofluorinated impurity of the latter [259]. No changes were observed by NMR when the drug was subjected to ionizing radiation (20–200 kGy) [260]. The degradation product of a fluorinated thiazole-containing compound was identified by extensive application of NMR spectroscopy, including 19F and 15N techniques [261].
Scheme 7.
Last step of the synthesis of fluconazole and generation of the monofluorinated impurity.
19F Solid state NMR spectroscopy was associated with chemometrics in order to study the solid phases of atorvastatin. The proposed method allows recognizing the mixtures of the prepared forms of the API with a LOD of the crystalline phase impurities in the amorphous phase < 1% [262]. In addition, the distinction between monomeric and dimeric fluorinated impurities was carried out by 19F NMR spectroscopy [263]. The complete assignment of a mixture of isomers of 1,3-perfluorodimethylcyclohexane as well as some impurities were identified using a combination of 19F-COSY, 19F-TOCSY and 19F-NOESY experiments[264].
15N NMR spectroscopy has very low sensitivity; however, it has been employed for confirming the identity of impurities and drug degradation products [260]. A cationic impurity was identified in preladenant employing a series of 1D and 2D NMR experiments, including the gHMBCAD experiment for accessing1H–15N long-range correlation information [265] and the structure of an impurity in valsartan was confirmed with the aid of 15N NMR experiments [266].
4. Conclusions and perspectives
The presence of minor amounts of unwanted chemicals might influence the efficacy and safety of the pharmaceutical products. Therefore, their structures should be elucidated when present at level higher than 0.1%. These impurities are also synthesized based on the suggested structures, as standards for selective analytical methods for their quantitation in drug substance and products.
NMR spectroscopy is finding increasing application for the identification of unknown organic impurities embedded in pharmaceutical preparations. In addition, this is the most suitable technique for the identification and the qualitative analysis of mixtures, where some of the compounds are unknown, and almost unsubstitutable for structural analysis of epimers, rotamers and even some cases of enantiomers. NMR is highly informative and it allows to draw conclusions on the structure of the components in a mixture of related compounds because the NMR spectrum is sensitive to even small modifications in their molecular geometry or bonding.
NMR spectroscopy is usually employed to obtain structural information. In some cases, it is used to confirm structural proposals made as a result of MS experiments, since the combination of NMR and MS is one of the most powerful approaches to acquire complete information on the structure of a studied molecule or mixture of compounds.
The quantitative determination of the level of impurities is one of the major applications of qNMR in this area. This can be done along with acquisition of their structural information. The determination of related impurities in a mixture, as well as content of residual solvent, isomeric composition, molar ratio and even the course of a degradation can be also achieved.
It has been shown that LC–NMR ranks among the most powerful hyphenated techniques for the separation and structure elucidation of complex organic molecules, also providing quantitative information. Due to the success of LC–NMR hyphenation, it can be foreseen that this approach will be extended to other multidimensional separation schemes in the near future. Despite the number of years of their evolution, coupled NMR techniques are still in their infancy and will require much more time to become established. Particularly, GC–NMR could be highly useful for volatiles. It is also expected that instrumental improvements will make more feasible the coupling of fractionation with 13C NMR detection.
The relatively low sensitivity, sometimes poor reproducibility and high instrumental costs are current substantial limitations of this spectroscopy. State-of-the-art NMR equipment, including higher magnetic fields and cryo-probes exhibit enhanced sensitivity. However, despite the latest advances, it should be stressed that current sensitivity still makes NMR unsuitable for quantitative analysis of most of the genotoxic impurities, even employing the most sensitive 1H NMR techniques.
Conquering all these technological frontiers required for trace and impurity analysis will open a completely new area of exploration and research in pharmaceutical analysis.
Acknowledgements
The authors gratefully acknowledge Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Secretaría de Ciencia y Técnica (SECyT-UNR) and Secretaría de Ciencia, Tecnología e Innovación (SECTeI) for financial support. NLC Thanks CONICET for her fellowship.
Appendix A. Acronyms, abbreviations and their definitions, regarding NMR experiments
| Acronyma/abbreviation | Meaning |
| 1D, 1D-NMR | Mono-dimensional NMR experiment |
| 1H qNMR, qHNMR | Quantitative proton NMR experiment |
| 2D, 2D-NMR | Two-dimensional NMR experiment |
| APT | Attached Proton Test |
| COSY | Homonuclear Correlation Spectroscopy |
| DBPPSTE DOSY | DOSY Bipolar Pulse Pair Stimulated Echo |
| DEPT | Distortionless Enhancement by Polarization Transfer |
| DOSY | Diffusion Ordered Spectroscopy |
| DQF-COSY | Double Quantum Filtered COSY |
| HETCOR | Heteronuclear Shift Correlation |
| HMBC | Heteronuclear Multiple-Bond Correlation |
| HMQC | Heteronuclear Multiple-Quantum Correlation |
| HSQC | Heteronuclear Single Quantum Correlation |
| NOE | Nuclear Overhauser Effect |
| NOESY | Nuclear Overhauser Effect Spectroscopy |
| qNMR | Quantitative NMR Spectroscopy |
| ROESY | Rotating-Frame NOE Spectroscopy |
| TOCSY | Total Correlation Spectroscopy |
| zTOCSY | z-Filtered Total Correlation Spectroscopy |
a
Prefix and suffix letters are added to more elaborate experiments. Letter ‘g’ is employed to denote the field gradient version of the experiment. The suffix ‘AD’ indicates the use of adiabatic pulses.
References
- [1]
- M. Sautel, A.M. Krstulovic
- Pharmaceutical analysis beyond compendial requirements
- J. Sep. Sci., 28 (28) (2005), pp. 1538–1629
- [2]
- S. Ahuja, K.M. Alsante (Eds.), Handbook of Isolation and Characterization of Impurities in Pharmaceuticals, Elsevier, Amsterdam, The Netherlands (2003)
- [3]
- S. Husain, R. Nageswara Rao
- Monitoring of process impurities in drugs
- Z. Deyl, I. Miksik, F. Tagliero, E. Tesarová (Eds.), Advanced Chromatographic and Electromigration Methods in Biosciences, Elsevier, Amsterdam, The Netherlands (1998), pp. 833–888
- Article
|
PDF (2179 K)
|
View Record in Scopus
| | |
Citing articles (1) - [4]
- K.M. Alsante, R.C. Friedmann, T.D. Hatajik, L.L. Lohr, T.R. Sharp, K.D. Snyder, E.J. Szczesny
- Degradation and impurity analysis for pharmaceutical drug candidates
- S. Ahuja, S. Scypinski (Eds.), Handbook of Modern Pharmaceutical Analysis, Academic Press, San Diego, USA (2001), pp. 85–172
- Article
|
PDF (5199 K)
|
View Record in Scopus
| | |
Citing articles (13) - [5]
- C. Todd, E. Sheinin
- S. Ahuja, M. Dong (Eds.), Handbook of Pharmaceutical Analysis by HPLC, Academic Press, San Diego, USA (2005), pp. 359–377
- View Record in Scopus
| | |
Citing articles (1) - [6]
- L.Z. Zhou
- S. Ahuja, M. Dong (Eds.), Handbook of Pharmaceutical Analysis by HPLC, Academic Press, San Diego, USA (2005), pp. 499–568
- Article
|
PDF (7837 K)
|
View Record in Scopus
| | |
Citing articles (3) - [7]
- J.C. Berridge
- Impurities in drug substances and drug products: new approaches to quantification and qualification
- J. Pharm. Biomed. Anal., 14 (1995), pp. 7–12
- Article
|
PDF (479 K)
|
View Record in Scopus
| | |
Citing articles (25) - [8]
- A.M. Krstulovic, C.R. Lee
- Defining drug purity through chromatographic and related methods: current status and perspectives
- J. Chromatogr. B, 689 (1997), pp. 137–153
- Article
|
PDF (859 K)
|
View Record in Scopus
| | |
Citing articles (20) - [9]
- R.N. Rao, V. Nagaraju
- An overview of the recent trends in development of HPLC methods for determination of impurities in drugs
- J. Pharm. Biomed. Anal., 33 (2003), pp. 335–377
- [10]
- J.V. Rompay
- Purity determination and evaluation of new drug substances
- J. Pharm. Biomed. Anal., 4 (1986), pp. 725–732
- [11]
- S. Ahuja
- Assuring quality of drugs by monitoring impurities
- Adv. Drug Deliv. Rev., 59 (2007), pp. 3–11
- Article
|
PDF (227 K)
|
View Record in Scopus
| | |
Citing articles (35) - [12]
- S. Ahuja
- Impurities Evaluation of Pharmaceutical
- Marcel Dekker, New York, USA (1998)
- [13]
- S. Görög
- Identification and Determination of Impurities in Drugs
- Elsevier, Amsterdam, The Netherlands (2000)
- [14]
- M. Webb, R. Smith (Eds.), Analysis of Drug Impurities, Blackwell, Oxford UK (2007)
- [15]
- N. Rahman, S.N. Hejaz Azmi, H.-F. Wu
- The importance of impurity analysis in pharmaceutical products: an integrated approach
- Accred. Qual. Assur., 11 (2006), pp. 69–74
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (11) - [16]
- D. Bartos, S. Görög
- Recent advances in the impurity profiling of drugs
- Curr. Pharm. Anal., 4 (2008), pp. 215–230
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (33) - [17]
- S.B.K.B. Bari, Y.S. Jaiswal, A.A. Shikhedkar
- Impurity profile: significance in active pharmaceutical ingredient
- Eurasian J. Anal. Chem., 2 (2007), pp. 32–53
- View Record in Scopus
| | |
Citing articles (32) - [18]
- B. De Spiegeleer, V. Vergote, A. Pezeshki, K. Peremans, C. Burvenich
- Impurity profiling quality control testing of synthetic peptides using liquid-chromatography-photodiode array fluorescence and liquid chromatography–electrospray ionization-mass spectrometry. The obestatin case
- Anal. Biochem., 376 (2008), pp. 229–234
- Article
|
PDF (277 K)
|
View Record in Scopus
| | |
Citing articles (36) - [19]
- S.W. Baertschi
- Analytical methodologies for discovering and profiling degradation-related impurities
- Trends Anal. Chem., 25 (2006), pp. 758–767
- Article
|
PDF (161 K)
|
View Record in Scopus
| | |
Citing articles (28) - [20]
- I.C.H.
- Impurities in new drug substances Q3A (R2)
- International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2006)
- [21]
- ICH
- Impurities in new drug products Q3B(R2)
- International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2006)
- [22]
- C. Foti, K. Alsante, G. Cheng, T. Zelesky, M. Zell
- Tools and workflow for structure elucidation of drug degradation products
- Trends Anal. Chem., 49 (2013), pp. 89–99
- Article
|
PDF (1269 K)
|
View Record in Scopus
| |
Citing articles (3) - [23]
- K. Wilczewska, A. Kot-Wasik, J. Namieśnik
- LC–MS and LC–NMR as complementary techniques for the determination of pharmaceuticals in dosage formulations
- Crit. Rev. Anal. Chem., 43 (2013), pp. 148–175
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (2) - [24]
- S. Singh, T. Handa, M. Narayanam, A. Sahu, M. Junwal, R.P. Shah
- A critical review on the use of modern sophisticated hyphenated tools in the characterization of impurities and degradation products
- J. Pharm. Biomed. Anal., 69 (2012), pp. 148–173
- Article
|
PDF (1599 K)
|
View Record in Scopus
| |
Citing articles (38) - [25]
- J. Chin, J.B. Fell, M. Jarosinski, M.J. Shapiro, J.R. Wareing
- HPLC/NMR in combinatorial chemistry
- J. Org. Chem., 63 (1998), pp. 386–390
- Full Text via CrossRef
- [26]
- H. Thiele, G. McLeod, M. Niemitz, T. Kühn
- Structure verification of small molecules using mass spectrometry and NMR spectroscopy
- Monatsh. Chem., 142 (2011), pp. 717–730
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5) - [27]
- S.K. Bharti, R. Roy
- Quantitative 1H NMR spectroscopy
- Trends Anal. Chem., 35 (2012), pp. 5–26
- Article
|
PDF (788 K)
|
View Record in Scopus
| |
Citing articles (40) - [28]
- M. Findeisen, S. Berger
- 50 and More Essential NMR Experiments: A Detailed Guide
- Wiley, New York, USA (2013)
- [29]
- S. Berger, S. Braun
- 200 and More NMR Experiments
- Wiley, New York, USA (2004)
- [30]
- G.A. Webb (Ed.), Modern Magnetic Resonance, Springer, Heidelberg, Germany (2007)
- [31]
- J. Keeler
- Understanding NMR Spectroscopy
- Wiley, New York, USA (2013)
- [32]
- H. Heise, S. Matthews (Eds.), Modern NMR Methodology, Springer, Heidelberg, Germany (2013)
- [33]
- N.E. Jacobsen
- NMR Spectroscopy Explained: Simplified Theory, Applications and Examples for Organic Chemistry and Structural Biology
- Wiley, New York, USA (2007)
- [34]
- M.Y. Iqbal, K.M.V. Narayana Rao, G. Sridhar, P. Padmanabha Raju, G.R. Deshpande, J.M. Babu
- Characterization and relative response factor determination of process related impurity in naproxen by nuclear magnetic resonance spectroscopy
- J. Pharm. Biomed. Anal., 56 (2011), pp. 484–490
- Article
|
PDF (846 K)
|
View Record in Scopus
| |
Citing articles (4) - [35]
- U. Holzgrabe, R. Deubner, C. Schollmayer, B. Waibel
- Quantitative NMR spectroscopy – applications in drug analysis
- J. Pharm. Biomed. Anal., 38 (2005), pp. 806–812
- Article
|
PDF (262 K)
|
View Record in Scopus
| |
Citing articles (74) - [36]
- M. Malet-Martino, U. Holzgrabe
- NMR techniques in biomedical and pharmaceutical analysis
- J. Pharm. Biomed. Anal., 55 (2011), pp. 1–15
- Article
|
PDF (753 K)
|
View Record in Scopus
| |
Citing articles (40) - [37]
- D.W.T. Claridge
- High Resolution NMR Techniques in Organic Chemistry
- Elsevier, Amsterdam, The Netherlands (2009)
- [38]
- S. Ito, T. Miki, M. Yoshikawa, M. Hamada, Y. Kawate, S. Hayashi, A. Sato, T. Kiyoshi, F. Matsumoto, H. Nagai, H. Wada, S. Fukui, T. Noguchi
- Test restults of long term operation of the superfluid cooled cryostat for a 1 GHz NMR Spectrometer
- IEEE Trans. Appl. Supercond., 12 (2002), pp. 1347–1350
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4) - [39]
- P.F. Flynn, D.L. Mattiello, H.D.W. Hill, A.J. Wand
- Optimal use of cryogenic probe technology in NMR studies of proteins
- J. Am. Chem. Soc., 122 (2000), pp. 4823–4824
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (65) - [40]
- H. Nagai, A. Sato, T. Kiyoshi, F. Matsumoto, H. Wada, S. Ito, T. Miki, M. Yoshikawa, Y. Kawate, S. Fukui
- Development and testing of superfluid-cooled 900 MHz NMR magnet
- Cryogenics, 41 (2001), pp. 623–630
- Article
|
PDF (779 K)
|
View Record in Scopus
| |
Citing articles (8) - [41]
- Y.Q. Song, B.M. Goodson, A. Pines
- NMR and MRI using laser-polarized xenon
- Spectroscopy, 14 (1999), pp. 26–33
- [42]
- U. Holzgrabe, B. Diehl, I. Wawer (Eds.), NMR Spectroscopy in Pharmaceutical Analysis, Elsevier, Amsterdam, The Netherlands (2008)
- [43]
- A. Karioti, E. Giocaliere, C. Guccione, G. Pieraccini, E. Gallo, A. Vannaccic, A.R. Bilia
- Combined HPLC–DAD–MS, HPLC–MSn and NMR spectroscopy for quality control of plant extracts: the case of a commercial blend sold as dietary supplement
- J. Pharm. Biomed. Anal., 88 (2014), pp. 7–15
- Article
|
PDF (2222 K)
|
View Record in Scopus
| |
Citing articles (3) - [44]
- I. McEwen, A. Elmsjö, A. Lehnström, A. Hakkarainen, B.M. Johansson
- Screening of counterfeit corticosteroid in creams and ointments by NMR spectroscopy
- J. Pharm. Biomed. Anal., 70 (2012), pp. 245–250
- Article
|
PDF (575 K)
|
View Record in Scopus
| |
Citing articles (6) - [45]
- T. Beyer, B. Diehl, U. Holzgrabe
- Quantitative NMR spectroscopy of biologically active substances and excipients
- Bioanal. Rev., 2 (2010), pp. 1–22
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (22) - [46]
- S. Görög, C. Szántay Jr.
- Spectroscopic methods in drug quality control and development
- J. Lindon, G. Tranter, D. Koppenaal (Eds.), Encyclopedia of Spectroscopy and Spectrometry (2nd ed.), vol. 3Elsevier, Oxford, UK (2010), pp. 2640–2650
- Article
|
PDF (537 K)
|
View Record in Scopus
| |
Citing articles (5) - [47]
- C. Szántay Jr., Z. Béni, G. Balogh, T. Gáti
- The changing role of NMR spectroscopy in off-line impurity identification: a conceptual view
- Trends Anal. Chem., 25 (2006), pp. 806–820
- Article
|
PDF (316 K)
|
View Record in Scopus
| |
Citing articles (21) - [48]
- C. Szántay Jr., A. Demeter
- NMR spectroscopy
- S. Görög (Ed.), Identification and Determination of Impurities in Drugs (1st ed.), Elsevier, Amsterdam, The Netherlands (2000), pp. 109–143
- [49]
- A. Aranyi
- Isolation of impurities by (semi)preparative HPLC
- S. Görög (Ed.), Identification and Determination of Impurities in Drugs (1st ed.), Elsevier, Amsterdam, The Netherlands (2000), pp. 240–251
- Article
|
PDF (537 K)
|
View Record in Scopus
| |
Citing articles (3) - [50]
- G.F. Pauli, T. Gödecke, B.U. Jaki, D.C. Lankin
- Quantitative 1H NMR. Development and potential of an analytical method: an update
- J. Nat. Prod., 75 (2012), pp. 834–851
- View Record in Scopus
| |
Citing articles (70) - [51]
- T. Ründlof, T.R. McEwen, M. Johansson, T. Arvidsson
- Use and qualification of primary and secondary standards employed in quantitative 1H NMR spectroscopy of pharmaceuticals
- J. Pharm. Biomed. Anal. (2013), p. 010 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.2013.09
- [52]
- M. Weber, C. Hellriegel, A. Rück, J. Wuethrich, P. Jenks
- Using high-performance 1H NMR (HP-qNMR®) for the certification of organic reference materials under accreditation guidelines – describing the overall process with focus on homogeneity and stability assessment
- J. Pharm. Biomed. Anal. (2013), p. 007 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.2013.09
- View Record in Scopus
| |
Citing articles (1) - [53]
- H. Jancke, F. Malz, W. Haesselbarth
- Structure analytical methods for quantitative reference applications
- Accred. Qual. Assur., 10 (2005), pp. 421–429
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (22) - [54]
- T. Schoenberger
- Determination of standard sample purity using the high-precision 1H-NMR process
- Anal. Bioanal. Chem., 403 (2012), pp. 247–254
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (14) - [55]
- F. Malz, H. Jancke
- Validation of quantitative NMR
- J. Pharm. Biomed. Anal., 38 (2005), pp. 813–823
- Article
|
PDF (335 K)
|
View Record in Scopus
| |
Citing articles (206) - [56]
- U. Holzgrabe
- Quantitative NMR spectroscopy in pharmaceutical applications
- Prog. Nucl. Magn. Reson. Spectrosc., 57 (2010), pp. 229–240
- Article
|
PDF (1009 K)
|
View Record in Scopus
| |
Citing articles (74) - [57]
- S. Mahajan, I.P. Singh
- Determining and reporting purity of organic molecules: why qNMR
- Magn. Reson. Chem., 51 (2013), pp. 76–81
- View Record in Scopus
| |
Citing articles (10) - [58]
- R. Nogueira, B.C. Garrido, R.M. Borges, G.E.B. Silva, S.M. Queiroz, V.S. Cunha
- Development of a new sodium diclofenac certified reference material using the mass balance approach and 1H qNMR to determine the certified property value
- Eur. J. Pharm. Sci., 48 (2013), pp. 502–513
- Article
|
PDF (940 K)
|
View Record in Scopus
| |
Citing articles (7) - [59]
- M. Weber, C. Hellriegel, A. Rück, R. Sauermoser, J. Wüthrich
- Using high-performance quantitative NMR (HP-qNMRW) for certifying traceable and highly accurate purity values of organic reference materials with uncertainties < 0.1%
- Accred. Qual. Assur., 18 (2013), pp. 91–98
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12) - [60]
- U. Holzgrabe
- Quantitative NMR spectroscopy in pharmaceutical applications
- Progr. NMR Spectrosc., 57 (2010), pp. 229–240
- Article
|
PDF (1009 K)
|
View Record in Scopus - [61]
- R. Deubner, U. Holzgrabe
- Micellar electrokinetic capillary chromatography, high performance liquid chromatography and nuclear magnetic resonance-three orthogonal methods for characterization of critical drugs
- J. Pharm. Biomed. Anal., 35 (2004), pp. 459–467
- Article
|
PDF (387 K)
|
View Record in Scopus
| | |
Citing articles (25) - [62]
- K. Albert (Ed.), On-Line LC–NMR and Related Techniques, Wiley, New York USA (2002)
- [63]
- M.V. Silva Elipe
- LC–NMR and Other Hyphenated NMR Techniques: Overview and Applications
- Wiley, Hoboken, USA (2012)
- [64]
- Y. Kazakevich, R. LoBrutto (Eds.), HPLC for Pharmaceutical Scientists, Wiley, New York, USA (2007), pp. 901–936
- [65]
- N.C. Gonella
- LC–NMR: Expanding the Limits of Structure Elucidation
- CRC, Boca Raton, USA (2013)
- [66]
- M.-H. Wann
- 20 Application of LC–NMR in pharmaceutical analysis
- Sep. Sci. Technol., 6 (2005), pp. 569–579
- Article
|
PDF (532 K)
|
View Record in Scopus
| | |
Citing articles (7) - [67]
- J.R. Kesting, K.T. Johansen, J.W. Jaroszewski
- Hyphenated NMR techniques
- A.J. Dingley, S.M. Pascal (Eds.), Advances in Biomedical Spectroscopy, Volume 3: Biomolecular NMR Spectroscopy, IOP Press, Amsterdam, The Netherlands (2011), pp. 413–434
- View Record in Scopus
| | |
Citing articles (4) - [68]
- M. Kühnle, D. Kreidler, H. Czesla, K. Holtin, P. Schuler, W. Schaal, V. Schurig, K. Albert
- On-line coupling of gas chromatography to nuclear magnetic resonance spectroscopy: a method for the analysis of volatile stereoisomers
- Anal. Chem., 80 (2008), pp. 5481–5486
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (18) - [69]
- M. Grynbaum, D. Kreidler, J. Rehbein, A. Purea, P. Schuler, W. Schaal, H. Czesla, A. Webb, V. Schurig, K. Albert
- Hyphenation of gas chromatography to microcoil 1H nuclear magnetic resonance spectroscopy
- Anal. Chem., 79 (2007), pp. 2708–2713
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (25) - [70]
- K. Pusecker, J. Schewitz, P. Gfrorer, L.-H. Tseng, K. Albert, E. Bayer, I.D. Wilson, N.J. Bailey, G.B. Scarfe, J.K. Nicholson, L.C. Lindon
- On-flow identification of metabolites of paracetamol from human urine using directly coupled CZE-NMR and CEC-NMR spectroscopy
- Anal. Commun., 35 (1998), pp. 213–215
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (41) - [71]
- J. Schewitz, P. Gfrorer, K. Pusecker, L.-H. Tseng, K. Albert, E. Bayer, I.D. Wilson, N.J. Bailey, G.B. Scarfe, J.K. Nicholson, J.C. Lindon
- Directly coupled CZE-NMR and CEC-NMR spectroscopy of metabolite analysis: paracetamol metabolites in human tissue
- Analyst, 12 (1998), pp. 2835–2837
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (44) - [72]
- K. Pusecker, J. Schewitz, P. Gfrorer, L.-H. Tseng, K. Albert, E. Bayer
- On-line coupling of capillary electrochromatography, capillary electrophoresis, and capillary HPLC with nuclear magnetic resonance spectroscopy
- Anal. Chem., 70 (1998), pp. 3280–3285
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (93) - [73]
- P. Gfrorer, L.-H. Tseng, E. Rapp, K. Albert, E. Bayer
- Influence of pressure upon coupling pressurized capillary electrochromatography with nuclear magnetic resonance spectroscopy
- Anal. Chem., 73 (2001), pp. 3234–3239
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (49) - [74]
- P. Gfrorer, J. Schewitz, K. Pusecker, L.-H. Tseng, K. Albert, E. Bayer
- Gradient elution capillary electrochromatogrpahy and hyphenation with nuclear magnetic resonance
- Electrophoresis, 20 (1999), pp. 3–8
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (51) - [75]
- M.J. Little, N. Aubry, M.-E. Beaudoin, N. Goudreau, S.R. LaPlante
- Quantifying trifluoroacetic acid as a counterion in drug discovery by 19F NMR and capillary electrophoresis
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1324–1330
- Article
|
PDF (187 K)
|
View Record in Scopus
| | |
Citing articles (14) - [76]
- D.A. Jayawickrama, J.V. Sweedler
- Hyphenation of capillary separations with nuclear magnetic resonance spectroscopy
- J. Chromatogr. A, 1000 (2003), pp. 819–840
- Article
|
PDF (1199 K)
|
View Record in Scopus
| | |
Citing articles (50) - [77]
- N. Wu, T.L. Peck, A.G. Webb, R.L. Magin, J.V. Sweedler
- Nanoliter volume sample cells for 1H NMR application to on-line detection in capillary electrophoresis
- J. Am. Chem. Soc., 116 (1994), pp. 7929–7930
- Full Text via CrossRef
- [78]
- J. Schewitz, R. Pusecker, P. Gfrorer, U. Gotz, L.-H. Tseng, K. Albert, E. Bayer
- Direct coupling of capillary electrophoresis and nuclear magnetic resonance spectroscopy for the identification of a dinucleotide
- Chromatographia, 50 (1999), pp. 333–337
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (27) - [79]
- M. Kühnle, D. Kreidler, K. Holtin, H. Czesla, P. Schuler, V. Schurig, K. Albert
- Online coupling of enantioselective capillary gas chromatography with proton nuclear magnetic resonance spectroscopy
- Chirality, 22 (2010), pp. 808–812
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (8) - [80]
- P. Novak, M. Cindric, P. Tepes, S. Dragojevic, M.K. Ilija
- Mihaljevic, Identification of impurities in acarbose by using an integrated liquid chromatography-nuclear magnetic resonance and liquid chromatography-mass spectrometry approach
- J. Sep. Sci., 28 (2005), pp. 1442–1447
- View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (11) - [81]
- F. Xu, A.J. Alexander
- The design of an on-line semi-preparative LC-SPE-NMR system for trace analysis
- Magn. Reson. Chem., 43 (2005), pp. 776–782
- View Record in Scopus
| | |
Citing articles (31) - [82]
- J.R. Kesting, J. Huang, D. Sorensen
- Identification of adulterants in a Chinese herbal medicine by LC-HRMS and LC-MS-SPE/NMR and comparative in vivo study with standards in a hypertensive rat model
- J. Pharm. Biomed. Anal., 51 (2010), pp. 705–711
- Article
|
PDF (344 K)
|
View Record in Scopus
| |
Citing articles (27) - [83]
- T. Murakami, N. Fukutsu, J. Kondo, T. Kawasaki, F. Kusu
- Application of liquid chromatography-two-dimensional nuclear magnetic resonance spectroscopy using pre-concentration column trapping and liquid chromatography–mass spectrometry for the identification of degradation products in stressed commercial amlodipine maleate tablets
- J. Chromatogr. A, 1181 (2008), pp. 67–76
- Article
|
PDF (1021 K)
|
View Record in Scopus
| |
Citing articles (22) - [84]
- J.K. Roberts, R.J. Smith
- Use of liquid chromatography-nuclear magnetic resonance spectroscopy for the identification of impurities in drug substances
- J. Chromatogr. A, 677 (1994), pp. 385–389
- Article
|
PDF (380 K)
|
View Record in Scopus
| |
Citing articles (44) - [85]
- I.N. Glazkov, N.L. Bochkareva, I.A. Revel’skii
- Determination of organic impurities in pharmaceutical preparations
- J. Anal. Chem., 60 (2005), pp. 107–118
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12) - [86]
- M. Babjak, G. Balogh, M. Gazdag, S. Gorog
- Estimation of impurity profiles of drugs and related materials. Part XXI. HPLC/UV/MS study of the impurity profile of ethynodiol diacetate
- J. Pharm. Biomed. Anal., 29 (2002), pp. 1153–1157
- Article
|
PDF (112 K)
|
View Record in Scopus
| |
Citing articles (14) - [87]
- F. Guo, H. Feng, Y. Wang, C. Zhang, Y. Li
- Characterization of related impurities in megestrol acetate
- J. Pharm. Biomed. Anal., 41 (2006), pp. 1418–1422
- Article
|
PDF (252 K)
|
View Record in Scopus
| |
Citing articles (4) - [88]
- J. Hammond, I. Jones
- The use of chromatography and online structure elucidation using spectroscopy
- R.J. Smith, M.L. Webb (Eds.), Analysis of Drug Impurities, Blackwell Publishing, Oxford, UK (2007)
- [89]
- N.P. Mishchenko, S.A. Fedoreev, V.P. Glazunov, V.A. Denisenko, N.P. Krasovskaya, L.I. Glebko, L.G. Maslov, P.S. Dmitrenok, V.L. Bagirova
- Isolation and identification of impurities in the parent substance of echinochrome and in the drug histochrome
- Pharm. Chem. J., 38 (2004), pp. 50–53
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4) - [90]
- V.V. Podgorskii, A.S. Mikhalev, G.A. Kalabin
- Quantitative NMR spectroscopy for quality control of drugs and pharmaceuticals
- Pharm. Chem. J., 45 (2011), pp. 194–197
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5) - [91]
- Y. Peng, J. Luo, Q. Lu, X. Chen, Y. Xie, L. Chen, W. Yang, S. Du
- HPLC analysis, semi-preparative HPLC preparation and identification of three impurities in salidroside bulk drug
- J. Pharm. Biomed. Anal., 49 (2009), pp. 828–832
- Article
|
PDF (357 K)
|
View Record in Scopus
| |
Citing articles (15) - [92]
- B. Wang, H. Wang, E. Wang, W. Yan, W. Ye, P. Li
- Isolation and structure characterization of related impurities in sodium tanshinone IIA sulfonate by LC/ESI-MSn and NMR
- J. Pharm. Biomed. Anal., 67–68 (2012), pp. 36–41
- Article
|
PDF (488 K)
|
View Record in Scopus
| |
Citing articles (3) - [93]
- Q.G. Zou, B.Y. Huang, T.T. He, P. Wei, Z.J. Zhang, P.K. Ouyang
- Identification, isolation, and characterization of impurities in sodium tanshinone IIA sulfonate, J
- Liq. Chromatogr. Relat. Technol., 32 (2009), pp. 2346–2360
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4) - [94]
- Z. Dubrovay, V. Háda, Z. Bénia, C. Szántay Jr.
- NMR and mass spectrometric characterization of vinblastine, vincristine and some new related impurities – Part I
- J. Pharm. Biomed. Anal., 84 (2013), pp. 293–308
- Article
|
PDF (1379 K)
|
View Record in Scopus
| |
Citing articles (4) - [95]
- V. Háda, Z. Dubrovay, A. Lakó-Futó, J. Galambos, Z. Gulyás, A. Aranyi, C. Szántay Jr.
- NMR and mass spectrometric characterization of vinblastine, vincristine and some new related impurities – Part II
- J. Pharm. Biomed. Anal., 84 (2013), pp. 309–322
- Article
|
PDF (1384 K)
|
View Record in Scopus - [96]
- X. Cao, Y. Tai, C. Sun, K. Wang, Y. Pan
- Characterization of impurities in semi-synthetic vinorelbine bitartrate by HPLC-MS with mass spectrometric shift technique
- J. Pharm. Biomed. Anal., 39 (2005), pp. 39–45
- Article
|
PDF (189 K)
|
View Record in Scopus
| |
Citing articles (4) - [97]
- K. Petritis, C. Elfakir, M. Dreux
- A comparative study of commercial liquid chromatographic detectors for the analysis of underivatized amino acids
- J. Chromatogr. A, 961 (2002), pp. 9–21
- Article
|
PDF (454 K)
|
View Record in Scopus
| |
Citing articles (108) - [98]
- U. Holzgrabe, C.J. Nap, T. Beyer, S. Almeling
- Alternatives to amino-acid-analysis for the purity control of pharmaceutical grade L-alanine
- J. Sep. Sci., 33 (2010), pp. 2402–2410
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12) - [99]
- S. Kopec, U. Holzgrabe
- Impurity profile of amino acids?
- Pharmeurope Sci. Notes, 17 (2005), pp. 39–45
- View Record in Scopus
| |
Citing articles (6) - [100]
- S. Rao Chitturi, C. Bharathia, A.V. Raghava Reddy, K. Chandrasekhar Reddy, H. Kumar Sharma, V. Kumar Handa, R. Dandala, V.H. Bindu
- Impurity profile study of lopinavir and validation of HPLC method for the determination of related substances in lopinavir drug substance
- J. Pharm. Biomed. Anal., 48 (2008), pp. 1430–1440
- [101]
- H. Liu, Z. Zhang, R.J. Linhardt
- Lessons learned from the contamination of heparin
- Nat. Prod. Rep., 26 (2009), pp. 313–321
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (132) - [102]
- S. Beni, J.F.K. Limtiaco, C.K. Larive
- Analysis and characterization of heparin impurities
- Anal. Bioanal. Chem., 399 (2011), pp. 527–539
- [SD-008]
- [103]
- USP
- Heparin Sodium, United States Pharmacopeia-National Formulary, USP 35-NF 30
- Rand McNally, Rockville, USA (2013), pp. 3040–3042 3403–3406
- [SD-008]
- [104]
- T. Beyer, M. Matz, D. Brinz, O. Rädler, B. Wolf, J. Norwig, K. Baumann, S. Alban, U. Holzgrabe
- Composition of OSCS-contaminated heparin occuring in 2008 in batches on the german market
- Eur. J. Pharm. Sci., 40 (2010), pp. 297–304
- [SD-008]
- [105]
- H. Ye, T.K. Toby, C.D. Sommers, H. Ghasriani, M.L. Trehy, W. Ye, R.E. Kolinski, L.F. Buhse, A. Al-Hakim, D.A. Keire
- Characterization of currently marketed heparin products: key tests for LMWH quality assurance
- J. Pharm. Biomed. Anal., 85 (2013), pp. 99–107
- [SD-008]
- [106]
- Q. Zang, D.A. Keire, R.D. Wood, L.F. Buhse, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
- Combining, (1)H NMR spectroscopy and chemometrics to identify heparin samples that may possess dermatan sulfate (DS) impurities or oversulfated chondroitin sulfate (OSCS) contaminants
- J. Pharm. Biomed. Anal., 54 (2011), pp. 1020–1029
- [SD-008]
- [107]
- P.A.J. Mourier, O.Y. Guichard, F. Herman, C. Viskov
- Heparin sodium compliance to USP monograph: structural elucidation of an atypical 2.18 ppm NMR signal
- J. Pharm. Biomed. Anal., 67–68 (2012), pp. 169–174
- [SD-008]
- [108]
- Q. Zang, D.A. Keire, L.F. Buhse, R.D. Wood, D.P. Mital, S. Haque, S. Srinivasan, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
- Identification of heparin samples that contain impurities or contaminants by chemometric pattern recognition analysis of proton NMR spectral data
- Anal. Bioanal. Chem., 401 (2011), pp. 939–955
- [SD-008]
- [109]
- Q. Zang, D.A. Keire, R.D. Wood, L.F. Buhse, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
- Determination of galactosamine impurities in heparin samples by multivariate regression analysis of their 1H NMR spectra
- Anal. Bioanal. Chem., 399 (2011), pp. 635–649
- [SD-008]
- [110]
- V. Ruiz-Calero, J. Saurina, M.T. Galceran, S. Hernández-Cassou, L. Puignou
- Potentiality of proton nuclear magnetic resonance and multivariate calibration methods for the determination of dermatan sulfate contamination in heparin samples
- Analyst, 125 (2000), pp. 933–938
- [SD-008]
- [111]
- V. Ruiz-Calero, J. Saurina, S. Hernández-Cassou, M.T. Galceran, L. Puignou
- Proton nuclear magnetic resonance characterization of glycosaminolgycans using chemometric techniques
- Analyst, 127 (2002), pp. 407–415
- [SD-008]
- [112]
- K.R. Riihinen, T. Gödecke, G.F. Pauli
- Purification of berry flavonol glycosides by long-bed gel permeation chromatography
- J. Chromatogr. A, 1244 (2012), pp. 20–27
- [SD-008]
- [113]
- F. Qiu, B. Friesen, J. McAlpine, G. Pauli
- Design of countercurrent separation of Ginkgo biloba terpene lactones by nuclear magnetic resonance
- J. Chromatogr. A, 1242 (2012), pp. 26–34
- [SD-008]
- [114]
- A. Tada, K. Takahashi, K. Ishizuki, N. Sugimoto, T. Suematsu, K. Arifuku, M. Tahara, T. Akiyama, Y. Ito, T. Yamazaki
- Absolute quantitation of stevioside and rebaudioside A in commercial standards by quantitative NMR
- Chem. Pharm. Bull., 61 (2013), pp. 33–38
- [SD-008]
- [115]
- V.G. Dongre, P.D. Ghugare, P. Karmuse, D. Singh, A. Jadhav, A. Kumar
- Identification and characterization of process related impurities in chloroquine and hydroxychloroquine by LC/IT/MS, LC/TOF/MS and NMR
- J. Pharm. Biomed. Anal., 49 (2009), pp. 873–879
- [SD-008]
- [116]
- B. Raman, B.A. Sharma, R. Butala, P.D. Ghugare, A. Kumar
- Structural elucidation of a process-related impurity in ezetimibe by LC/MS/MS and NMR
- J. Pharm. Biomed. Anal., 52 (2010), pp. 73–78
- [SD-008]
- [117]
- S. Guntupalli, U. Kumar Raya, N. Muralia, P. Badrinadh Gupta, V.J. Kumar, D. Satheesha, A. Islama
- Identification, isolation and characterization of process related impurities in ezetimibe
- J. Pharm. Biomed. Anal., 88 (2014), pp. 385–390
- [SD-008]
- [118]
- R. Nageswara Rao, K. Pawan, A. Maurya, Narasa Raju
- Isolation and characterization of a potential process related impurity of phenazopyridine HCl by preparative HPLC followed by MS–MS and 2D-NMR spectroscopy
- J. Pharm. Biomed. Anal., 49 (2009), pp. 1287–1291
- [SD-008]
- [119]
- D.V.N. Srinivasa Rao, N. Srinivas, Ch. Bharathi, Ch.S. Prasad, A. Ramesh Dandala, Naidu
- Identification and characterization of new impurity in didanosine
- J. Pharm. Biomed. Anal., 45 (2007), pp. 516–520
- [SD-008]
- [120]
- G. Madhusudhan Reddy, B. Vijaya Bhaskar, P. Pratap Reddy, S. Ashok, P. Sudhakar, J.M. Babu, K. Vyas, K. Mukkanti
- Structural identification and characterization of potential impurities of pantoprazole sodium
- J. Pharm. Biomed. Anal., 45 (2007), pp. 201–210
- [SD-008]
- [121]
- G. Madhusudhan Reddy, B. Vijaya Bhaskar, P. Pratap Reddy, P. Sudhakar, J. Moses Babu, K. Vyas, P. Ramachandra Reddy, K. Mukkanti
- Identification and characterization of potential impurities of rabeprazole sodium
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1262–1269
- View Record in Scopus
- [122]
- E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, S. Serra
- Impurities of tazarotene: isolation and structural characterization
- J. Pharm. Biomed. Anal., 46 (2008), pp. 574–576
- Article
|
PDF (180 K)
|
View Record in Scopus - [123]
- H. Zhou, Y. Tai, C. Sun, Y. Pan
- Separation and characterization of synthetic impurities of triclabendazole by reversed-phase high-performance liquid chromatography/electrospray ionization mass spectrometry
- J. Pharm. Biomed. Anal., 37 (2005), pp. 97–107
- Article
|
PDF (671 K)
|
View Record in Scopus - [124]
- G. Francese, F. Corana, O. Meneghetti, F. Marazza
- LC–MS characterization of trace impurities contained in calcium folinate
- J. Pharm. Biomed. Anal., 39 (2005), pp. 757–763
- Article
|
PDF (437 K)
|
View Record in Scopus - [125]
- C. Bharathi, C.S. Prasad, D. Vijaya Bharathi, R. Shankar, V. Janardhana Rao, R. Dandala, A. Naidu
- Structural identification and characterization of impurities in ceftizoxime sodium
- J. Pharm. Biomed. Anal., 43 (2007), pp. 733–740
- Article
|
PDF (489 K)
|
View Record in Scopus - [126]
- V. Srinivasan, H. Sivaramakrishnan, B. Karthikeyan
- Detection, isolation and characterization of principle synthetic route indicative impurity in telmisartan
- Arab. J. Chem. (2012), p. 022 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.arabjc.2012.03
- [127]
- A. Sampath, A. Raghupathi Reddy, B. Yakambaram, A. Thirupathi, M. Prabhakar, P. Pratap Reddy, V. Prabhakar Reddy
- Identification and characterization of potential impurities of valsartan, AT1 receptor antagonist
- J. Pharm. Biomed. Anal., 50 (2009), pp. 405–412
- Article
|
PDF (925 K)
|
View Record in Scopus - [128]
- B. Raman, B.A. Sharma, G. Mahale, D. Singh, A. Kumar
- Investigation and structural elucidation of a process related impurity in candesartan cilexetil by LC/ESI-ITMS and NMR
- J. Pharm. Biomed. Anal., 56 (2011), pp. 256–263
- Article
|
PDF (421 K)
|
View Record in Scopus - [129]
- M. Vuletic, M. Cindric, J.D. Koruznjak
- Identification of unknown impurities in simvastatin substance and tablets by liquid chromatography/tandem mass spectrometry
- J. Pharm. Biomed. Anal., 37 (2005), pp. 715–721
- Article
|
PDF (236 K)
|
View Record in Scopus - [130]
- R.M. Bianchini, P.M. Castellano, T.S. Kaufman
- Development and validation of an HPLC method for the determination of process-related impurities in pridinol mesylate, employing experimental designs
- Anal. Chim. Acta, 654 (2009), pp. 141–147
- Article
|
PDF (438 K)
|
View Record in Scopus - [131]
- T. Saji, P. Saroj Kumar, J. Subhash Chandra, K. Vineet, A. Ashutosh, V. Dharam
- Identification, synthesis and characterization of an unknown process related impurity in eslicarbazepine acetate active pharmaceutical ingredient by LC/ESI-IT/MS, 1H, 13C and 1H-1H COSY NMR
- J. Pharm. Anal., 86 (2013), pp. 204–213
- View Record in Scopus
- [132]
- X. Wang, H. Zhou, J. Zheng, C. Huang, W. Liu, L. Yu, S. Zeng
- Identification and characterization of four process-related impurities in retigabine
- J. Pharm. Biomed. Anal., 71 (2012), pp. 148–151
- Article
|
PDF (540 K)
|
View Record in Scopus - [133]
- D. Cui, Y. Li, M. Lian, F. Yang, Q. Meng
- Development of a simple and stability-indicating RP–HPLC method for determining olanzapine and related impurities generated in the preparative process
- Analyst, 136 (2011), pp. 3149–3156
- View Record in Scopus
|
Full Text via CrossRef - [134]
- G. Goverdhan, A. Raghupathi Reddy, K. Srinivas, V. Himabindu, G. Mahesh Reddy
- Identification, characterization and synthesis of impurities of zafirlukast
- J. Pharm. Biomed. Anal., 49 (2009), pp. 895–900
- Article
|
PDF (531 K)
|
View Record in Scopus - [135]
- M. Saravanan, K. Siva kumari, P. Pratap Reddy, M.N. Naidu, J. Moses Babu, A.K. Srivastava, T. Lakshmi Kumar, B.V.V.N. Chandra Sekhar, B. Satyanarayana
- Identification, synthesis, isolation and spectral characterization of potential impurities of montelukast sodium
- J. Pharm. Biomed. Anal., 48 (2008), pp. 708–715
- Article
|
PDF (408 K)
|
View Record in Scopus - [136]
- V. Shinde, A. Trivedi, P.R. Upadhayay, N.L. Gupta, D.G. Kanase, R. Chikate
- Identification of a new impurity in lisinopril
- J. Pharm. Biomed. Anal., 43 (2007), pp. 381–386
- Article
|
PDF (279 K)
|
View Record in Scopus - [137]
- V.R. Shinde, A. Trivedi, P.R. Upadhayay, N.L. Gupta, D.G. Kanase, R.C. Chikate
- Isolation and characterization of benazepril unknown impurity by chromatographic and spectroscopic methods
- J. Pharm. Biomed. Anal., 42 (2006), pp. 395–399
- Article
|
PDF (252 K)
|
View Record in Scopus - [138]
- M. Dousa, P. Gibala, S. Rádl, O. Klecán, Z. Mandelová, J. Brichác
- Identification, preparation and UHPLC determination of process-related impurity in zolmitriptan
- J. Pharm. Biomed. Anal., 58 (2012), pp. 1–6
- Article
|
PDF (170 K)
|
View Record in Scopus - [139]
- T.J. Sunder Raj, Ch. Bharathi, M. Saravana Kumar, P. Joseph Prabahar, H. Naveen Kumar, Kumar Sharma, Kalpesh Parikh
- Identification, isolation and characterization of process-related impurities in rizatriptan benzoate
- J. Pharm. Biomed. Anal., 49 (2009), pp. 156–162
- [140]
- E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, F. Sala
- Isolation and characterisation of impurities in adapalene
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1161–1163
- Article
|
PDF (140 K)
|
View Record in Scopus - [141]
- Y. Liu, L. Chen, Y. Ji
- Quantification and structural elucidation of potential impurities in agomelatine active pharmaceutical ingredient
- J. Pharm. Biomed. Anal., 81–82 (2013), pp. 193–201
- Article
|
PDF (1403 K)
|
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (3) - [142]
- B. Raman, B.A. Sharma, P.D. Ghugare, P.P. Karmuse, A. Kumar
- Semipreparative isolation and structural elucidation of an impurity in citalopram by LC/MS/MS
- J. Pharm. Biomed. Anal., 50 (2009), pp. 377–383
- Article
|
PDF (809 K)
|
View Record in Scopus
| |
Citing articles (13) - [143]
- B. Raman, B.A. Sharma, P.D. Ghugare, S. Nandavadekar, D. Singh, P.K. Karmuse, A. Kumar
- Structural elucidation of process-related impurities in escitalopram by LC/ESI-MS and NMR
- J. Pharm. Biomed. Anal., 53 (2010), pp. 895–901
- Article
|
PDF (283 K)
|
View Record in Scopus
| |
Citing articles (9) - [144]
- Y. Lin, P. Huang, H. Qi, L. Chen, D. Wang
- Isolation, synthesis and structure confirmation of the impurity in crude roflumilast product
- Res. Chem. Intermed., 39 (2013), pp. 3111–3115
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (2) - [145]
- S. Thomas, S. Kumar Paul, S. Shandilya, A. Agarwal, N. Saxena, A. Kumar Awasthi, H.B. Matta, D. Vir, C.S. Mathela
- Identification and structural elucidation of two process impurities and stress degradants in darifenacin hydrobromide active pharmaceutical ingredient by LC-ESI/MSn
- Analyst, 137 (2012), pp. 3571–3582
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (3) - [146]
- P. Elkins, D. Coleman, J. Burgess, M. Gardner, J. Hines, B. Scott, M. Kroenke, J. Larson, M. Lightner, G. Turner, J. White, P. Liu
- Characterization of the isomeric configuration and impurities of (Z)-endoxifen by 2D NMR, high resolution LC–MS, and quantitative HPLC analysis
- J. Pharm. Biomed. Anal., 88 (2014), pp. 174–179
- Article
|
PDF (507 K)
|
View Record in Scopus
| |
Citing articles (3) - [147]
- G.R. Bedford, D.N. Richardson
- Preparation and identification of cis and trans isomers of a substituted triarylethylene
- Nature, 5063 (1966), pp. 733–734
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (41) - [148]
- R.P. Shah, A. Sahu, S. Singh
- Identification and characterization of geometrical isomeric photo-degradation product of eprosartan using LC–MS and LC–NMR
- Eur. J. Chem., 2 (2011), pp. 152–157
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5) - [149]
- M.A. Khan, S. Sinha, S. Vartak, A. Bhartiya, S. Kumar
- LC determination of glimepiride and its related impurities
- J. Pharm. Biomed. Anal., 39 (2005), pp. 928–943
- Article
|
PDF (372 K)
|
View Record in Scopus
| |
Citing articles (31) - [150]
- S. Bouabdallah, H. Trabelsi, T. Ben Dhia, S. Sabbah, K. Bouzouita, R. Khaddar
- RP–HPLC and NMR study of cis–trans isomerization of enalaprilat
- J. Pharm. Biomed. Anal., 31 (2003), pp. 731–741
- Article
|
PDF (272 K)
|
View Record in Scopus
| |
Citing articles (16) - [151]
- V.G. Dongre, P.P. Karmuse, M.M. Nimbalkar, D. Singh, A. Kumar
- Application of GC–EI-MS for the identification and investigation of positional isomer in primaquine, an antimalarial drug
- J. Pharm. Biomed. Anal., 39 (2005), pp. 111–116
- Article
|
PDF (285 K)
|
View Record in Scopus
| |
Citing articles (15) - [152]
- V.G. Dongre, P.P. Karmuse, P.D. Ghugare, M. Gupta, B. Nerurkar, Ch. Shaha, A. Kumar
- Characterization and quantitative determination of impurities in piperaquine phosphate by HPLC and LC/MS/MS
- J. Pharm. Biomed. Anal., 43 (2007), pp. 186–195
- Article
|
PDF (1110 K)
|
View Record in Scopus
| |
Citing articles (10) - [153]
- N. Lindegardh, F. Giorgi, B. Galletti, M. Di Mattia, M. Quaglia, D. Carnevale, N.J. White, A. Mazzanti, N.P.J. Day
- Identification of an isomer impurity in piperaquine drug substance
- J. Chromatogr. A, 1135 (2006), pp. 166–169
- Article
|
PDF (243 K)
|
View Record in Scopus
| |
Citing articles (5) - [154]
- N. Fukutsu, T. Kawasaki, K. Saito, H. Nakazawa
- Application of high-performance liquid chromatography hyphenated techniques for identification of degradation products of cefpodoxime proxetil
- J. Chromatogr. A, 1129 (2006), pp. 153–159
- Article
|
PDF (473 K)
|
View Record in Scopus
| |
Citing articles (28) - [155]
- K.V.V. Prasada Rao, A. Rani, A.V. Raghava Reddy, C.H. Bharathi, R. Dandala, A. Naidu
- Isolation, structural elucidation and characterization of impurities in cefdinir
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1476–1482
- [156]
- C.A. Beasley, T.-L. Hwang, K. Fliszar, A. Abenda, D.G. McCollum, R.A. Reed
- Identification of impurities in ivermectin bulk material by mass spectrometry and NMR
- J. Pharm. Biomed. Anal., 41 (2006), pp. 1124–1134
- Article
|
PDF (320 K)
|
View Record in Scopus
| |
Citing articles (4) - [157]
- W. Liu, A. Zhou, F. Feng, H. Zhang, J. Zhou, W. Huang
- Qualitative and quantitative studies on impurities in G004, a potential hypoglycaemic agent, using liquid chromatography, nuclear magnetic resonance and mass spectrometry
- J. Pharm. Biomed. Anal., 56 (2011), pp. 627–632
- Article
|
PDF (473 K)
|
View Record in Scopus
| |
Citing articles (4) - [158]
- H. Wang, Z. Zhang, F. Xiong, L. Wu, P. Li, W. Ye
- Isolation and structure characterization of related impurities in etimicin sulfate by LC/ESI-MSn and NMR
- J. Pharm. Biomed. Anal., 55 (2011), pp. 902–907
- Article
|
PDF (364 K)
|
View Record in Scopus
| |
Citing articles (6) - [159]
- E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, L. Malpezzi
- Isolation and characterisation of a phenolic impurity in a commercial sample of duloxetine
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1573–1575
- Article
|
PDF (131 K)
|
View Record in Scopus
| |
Citing articles (20) - [160]
- S.H. Kwak, J.M. Seo, K.-I. Lee
- A facile asymmetric synthesis of (S)-duloxetine
- ARKIVOC, x (2010), pp. 55–61
- View Record in Scopus
| |
Citing articles (1) - [161]
- ICH
- Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical Substances, Guideline Q6A
- International Conference on Harmonisation, IFPMA, Geneva, Switzerland (1999)
- [162]
- D.W. Armstrong, J.T. Lee, L.W. Chang
- Enantiomeric impurities in chiral catalysts, auxiliaries and synthons used in enantioselective synthesis
- Tetrahedron: Asymmetry, 9 (1998), pp. 2043–2064
- Article
|
PDF (1020 K)
|
View Record in Scopus
| |
Citing articles (33) - [163]
- D.W. Armstrong, L.F. He, T. Yu, J.T. Lee, Y.S. Liu
- Enantiomeric impurities in chiral catalysts, auxiliaries, synthons and resolving agents. Part 2
- Tetrahedron: Asymmetry, 10 (1999), pp. 37–60
- Article
|
PDF (1094 K)
|
View Record in Scopus
| |
Citing articles (32) - [164]
- K. Huang, Z.S. Breitbach, D.W. Armstrong
- Enantiomeric impurities in chiral synthons, catalyst and auxiliaries. Part 3
- Tetrahedron: Asymmetry, 17 (2006), pp. 2821–2832
- Article
|
PDF (324 K)
|
View Record in Scopus
| |
Citing articles (12) - [165]
- R.J. Lewis, M.A. Bernstein, H.-F. Chang, D. Chapman, N. Pemberton
- Enantiomeric purity determination by NMR: proving the purity of a single enantiomer
- Tetrahedron: Asymmetry, 24 (2013), pp. 866–870
- Article
|
PDF (723 K)
|
View Record in Scopus
| |
Citing articles (3) - [166]
- T.J. Wenzel
- Discrimination of Chiral Compounds Using NMR Spectroscopy
- Wiley, Hoboken, USA (2007)
- [167]
- T.J. Wenzel, C.D. Chisholm
- Using NMR spectroscopic methods to determine enantiomeric purity and assign absolute stereochemistry
- Prog. Nucl. Magn. Reson. Spectrosc., 59 (2011), pp. 1–63
- Article
|
PDF (14543 K)
|
View Record in Scopus
| |
Citing articles (54) - [168]
- A. Berthod (Ed.), Chiral Recognition in Separation Methods, Springer, Berlin, Germany (2010)
- [169]
- G. Tarkanyi
- Quantitative approach for the screening of cyclodextrins by nuclear magnetic resonance spectroscopy in support of chiral separations in liquid chromatography and capillary electrophoresis. Enantioseparation of norgestrel with α-, β- and γ-cyclodextrins
- J. Chromatogr. A, 961 (2002), pp. 257–276
- Article
|
PDF (1306 K)
|
View Record in Scopus
| |
Citing articles (30) - [170]
- E. Yashima, C. Yamamoto, Y. Okamoto
- NMR studies of chiral discrimination relevant to the liquid chromatographic enantioseparation by a cellulose phenylcarbamate derivative
- J. Am. Chem. Soc., 118 (1996), pp. 4036–4048
- View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (155) - [171]
- M. Kagawa, Y. Machida, H. Nishi, J. Haginaka
- Enantiomeric purity determination of acetyl-l-carnitine by NMR with chiral lanthanide shift reagents
- J. Pharm. Biomed. Anal., 38 (2005), pp. 918–923
- Article
|
PDF (131 K)
|
View Record in Scopus
| |
Citing articles (8) - [172]
- D. Vi Ji, Y.-F. Liu, Q.-H. Wang, C.-F. Zhang, Z.-L. Yang
- Isolation, structure characterization and quantification of related impurities in asperosaponin
- Chin. J. Nat. Med., 11 (2013), pp. 419–426
- [173]
- R. Vasu Dev, J. Moses Babu, K. Vyasa, P. Sai Ram, P. Ramachandra, N.M. Sekhar, D.N. Mohan Reddy, N. Srinivasa Rao
- Isolation and characterization of impurities in docetaxel
- J. Pharm. Biomed. Anal., 40 (2006), pp. 614–622
- Article
|
PDF (560 K)
|
View Record in Scopus
| |
Citing articles (26) - [174]
- D. Kumar, R. Singh Tomar, S. Kumar Deolia, M. Mitra, R. Mukherjee, A.C. Burman
- Isolation and characterization of degradation impurities in docetaxel drug substance and its formulation
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1228–1235
- Article
|
PDF (250 K)
|
View Record in Scopus
| |
Citing articles (22) - [175]
- W. Xu, X. Jia, W. Liu, X. Zhang, Y. Chen, L. Zhou, S. You
- Identification and characterization of three impurities in clocortolone pivalate
- J. Pharm. Biomed. Anal., 62 (2012), pp. 167–171
- Article
|
PDF (485 K)
|
View Record in Scopus
| |
Citing articles (3) - [176]
- European Medicines Agency
- Committee for Medicinal Products for Human Use, Guideline on the Limits of Genotoxic Impurities, London, UK
- (2004)
- [177]
- C. Bharathi, P. Jayaram, J. Sunder Raj, M. Saravana Kumar, V. Bhargavi, V.K. Handa, R. Dandala, A. Naidu
- Identification, isolation and characterization of impurities of clindamycin palmitate hydrochloride
- J. Pharm. Biomed. Anal., 48 (2008), pp. 1211–1218
- Article
|
PDF (536 K)
|
View Record in Scopus
| |
Citing articles (8) - [178]
- Genotoxic Impurities in Pharmaceutical Products Regulations and Analysis, Agilent Technologies, Doc. 5991–1876.
- [179]
- A. Teasdale (Ed.), Genotoxic impurities: Strategies for Identification and Control, Wiley, New York, USA (2011)
- [180]
- Validation of quantitative NMR, Implications for early phase of pharmaceutical development, S. de Goeij, MSc. Thesis, Universiteit van Amsterdam, Amsterdam, The Netherlands, 2012.
- [181]
- I. Raid, Application of qNMR for methanesulfonic acid analysis in pharmaceutical industry, Poster 1180-5. Pittcon 2012, Pittsburgh, USA, March 13, 2012. http://ca.pittcon.org/Technical+Program/tpabstra12.nsf/Agenda+Time+Slots+Web/FC5ADE1BCFBE8CBD852578DA00608AAA?Opendocument (accessed 15.12.13).
- [182]
- D.P. Elder, D. Snodin, A. Teasdale
- Control and analysis of hydrazine, hydrazides and hydrazones—genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products
- J. Pharm. Biomed. Anal., 54 (2011), pp. 900–910
- Article
|
PDF (272 K)
|
View Record in Scopus - [183]
- P. Kovarikova, K. Vavrova, K. Tomalova, M. Schongut, K. Hruskova, P. Hruskova, J. Klimes
- HPLC–DAD and MS/MS analysis of novel drug candidates from the group of aromatic hydrazones revealing the presence of geometrical isomers
- J. Pharm. Biomed. Anal., 48 (2008), pp. 295–302
- Article
|
PDF (273 K)
|
View Record in Scopus - [184]
- S. Provera, L. Martini, G. Guercio, L. Turco, L. Costa, C. Marchioro
- Application of LC–NMR and HR-NMR to the characterization of biphenyl impurities in the synthetic route development for vestipitant, a novel NK1 antagonist
- J. Pharm. Biomed. Anal., 53 (2010), pp. 389–395
- Article
|
PDF (315 K)
|
View Record in Scopus - [185]
- K.W. Sigvardson, S.P. Adams, T. Bradford Barnes, K.F. Blom, J.M. Fortunak, M.J. Haas, K.L. Reilly, A.J. Repta, G.A. Nemeth
- The isolation and identification of a toxic impurity in XP315 drug substance
- J. Pharm. Biomed. Anal., 27 (2002), pp. 327–334
- Article
|
PDF (154 K)
|
View Record in Scopus - [186]
- I.C.H.
- Impurities Guideline for residual solvents Q3C(R5)
- International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2011)
- [187]
- C. B’Hymer
- Residual solvent testing: a review of gas-chromatographic and alternative techniques
- Pharm. Res., 20 (2003), pp. 337–343
- [188]
- M. Guerrini, A. Bisio, G. Torri
- Combined quantitative 1H and 13C nuclear magnetic resonance spectroscopy for characterization of heparin preparations
- Semin. Thromb. Hemost., 27 (2001), pp. 473–482
- View Record in Scopus
|
Full Text via CrossRef - [189]
- F. Zhang, B. Yang, M. Ly, K. Solakyildirim, Z. Xiao, Z. Wang, J.M. Beaudet, A.Y. Torelli, J.S. Dordick, R.J. Linhardt
- Structural characterization of heparins from different commercial sources
- Anal. Bioanal. Chem., 401 (2011), pp. 2793–2803
- View Record in Scopus
|
Full Text via CrossRef - [190]
- I. McEwen, A. Amini, I.M. Olofsson
- Identification and purity test of heparin by NMR. A summary of 2 years’ experience at the medical products agency
- Pharmaeuropa Bio Sci. Notes, 1 (2010), pp. 65–72
- View Record in Scopus
- [191]
- G.A. Neville, F. Mori, K.R. Holme, A.S. Perlin
- Monitoring the purity of pharmaceutical heparin preparations by high-field 1HNMR magnetic resonance spectroscopy
- J. Pharm. Sci., 78 (1989), pp. 101–104
- View Record in Scopus
|
Full Text via CrossRef - [192]
- M.D. Argentine, P.K. Owens, B.A. Olsen
- Strategies for the investigation and control of process-related impurities in drug substances
- Adv. Drug Deliv. Rev., 59 (2007), pp. 12–28
- Article
|
PDF (841 K)
|
View Record in Scopus - [193]
- P.F. Bross, R. Kane, A.T. Farrell, S. Abraham, K. Benson, M.E. Brower, S. Bradley, J.V. Gobburu, A. Goheer, S.L. Lee, J. Leighton, C.Y. Liang, R.T. Lostritto, W.D. McGuinn, D.E. Morse, A. Rahman, L.A. Rosario, S.L. Verbois, G. Williams, Y.C. Wang, R. Pazdur
- Approval summary for bortezomib for injection in the treatment of multiple myeloma
- Clin. Cancer Res., 10 (2004), pp. 3954–3964
- View Record in Scopus
|
Full Text via CrossRef - [194]
- S.R. Byrn, P.A. Tishmack, M.J. Milton, H. van de Velde
- Analysis of two commercially available bortezomib products: differences in assay of active agent and impurity profile
- AAPS PharmSciTech, 12 (2011), pp. 461–467
- View Record in Scopus
|
Full Text via CrossRef - [195]
- J. Forshed, B. Erlandsson, S.P. Jacobsson
- Quantification of aldehyde impurities in poloxamer by 1H NMR spectrometry
- Anal. Chim. Acta, 552 (2005), pp. 160–165
- Article
|
PDF (236 K)
|
View Record in Scopus - [196]
- C. Thomasin, P. Johansen, R. Alder, R. Besmsel, G. Hottinger, H. Altorfer, A.D. Wright, G. Wehrli, H.P. Merkle, B. Gander
- A contribution to overcoming the problems of residual solvents in biodegradable microspheres prepared by coacervation
- J. Pharm. Biopharm., 42 (1996), pp. 16–24
- View Record in Scopus
- [197]
- H.W. Avdovich, M.J. Lebelle, C. Savard, W.L. Wilson
- Nuclear magnetic resonance identification of solvent residue in cocaine
- Forensic Sci. Int., 49 (1991), pp. 225–235
- Article
|
PDF (690 K)
|
View Record in Scopus - [198]
- R.J. Mumper, M. Jay
- Poly(l-lactic acid) microspheres containing neutron-activatable holmium-165: a study of the physical characteristics of microspheres before and after irradiation in a nuclear reactor
- Pharm. Res., 9 (1992), pp. 149–154
- View Record in Scopus
|
Full Text via CrossRef - [199]
- D. Jain, P.K. Basniwal
- A forced degradation and impurity profiling: recent trends in analytical perspectives
- J. Pharm. Biomed. Anal., 86 (2013), pp. 11–35
- Article
|
PDF (1423 K)
|
View Record in Scopus - [200]
- S. Singh, M. Junwal, G. Modhe, H. Tiwari, M. Kurmi, N. Parashar, P. Sidduri
- Forced degradation studies to assess the stability of drugs and products
- Trends Anal. Chem., 49 (2013), pp. 71–88
- Article
|
PDF (520 K)
|
View Record in Scopus - [201]
- A. Awasthi, M. Razzak, R. Al-Kassas, D.R. Greenwood, J. Harvey, S. Garg
- Separation and identification of degradation products in eprinomectin formulation using LC, LTQ FT-MS, H/D exchange, and NMR
- J. Pharm. Biomed. Anal., 63 (2012), pp. 62–73
- Article
|
PDF (1484 K)
|
View Record in Scopus - [202]
- R.N. Rao, K. Ramakrishna, B. Sravan, K. Santhakumar
- RP–HPLC separation and ESI-MS, 1H, and 13C NMR characterization of forced degradants including process related impurities of carisbamate: method development and validation
- J. Pharm. Biomed. Anal., 77 (2013), pp. 49–54
- [SD-008]
- [203]
- A. Mohan, M. Hariharan, E. Vikraman, G. Subbaiah, B.R. Venkataraman, D. Saravanan
- Identification and characterization of a principal oxidation impurity in clopidogrel drug substance and drug product
- J. Pharm. Biomed. Anal., 47 (2008), pp. 183–189
- [SD-008]
- [204]
- R.M. Bianchini, P.M. Castellano, T.S. Kaufman
- Validated stability-indicating HPLC method for the determination of pridinol mesylate. Kinetics study of its degradation in acid medium
- J. Pharm. Biomed. Anal., 48 (2008), pp. 1151–1160
- [SD-008]
- [205]
- S. Doddaga, R. Peddakonda
- Chloroquine-N-oxide, a major oxidative degradation product of chloroquine: identification, synthesis and characterization
- J. Pharm. Biomed. Anal., 81–82 (2013), pp. 118–125
- [SD-008]
- [206]
- T. Dyakonov, A. Muir, H. Nasri, D. Toops, A. Fatmi
- Isolation and characterization of cetirizine degradation product: mechanism of cetirizine oxidation
- Pharm. Res., 27 (2010), pp. 1318–1324
- [SD-008]
- [207]
- C. Ashok, M. Golak, D. Adwait, V. Krishna, A. Himani, P. Umesh, G. Amol, S. Srinivas, M. Sharad, J. Deepali, C. Atul
- Isolation and structural elucidation of two impurities from a diacerein bulk drug
- J. Pharm. Biomed. Anal., 49 (2009), pp. 525–528
- [SD-008]
- [208]
- B.V. Subbaiah, K.K.S. Ganesh, G.V. Krishna, K. Vyas, R.V. Dev, K.S. Reddy
- Isolation and characterisation of degradant impurities in dipyridamole formulation
- J. Pharm. Biomed. Anal., 61 (2012), pp. 256–264
- [SD-008]
- [209]
- P. Novak, P. Tepes, M. Ilijas, I. Fistric, I. Bratos, A. Avdagic, Z. Hameršak, V.G. Marković, M. Dumić
- LC–NMR and LC–MS identification of an impurity in a novel antifungal drug icofungipen
- J. Pharm. Biomed. Anal., 50 (2009), pp. 68–72
- [SD-008]
- [210]
- C. Sun, J. Wu, D. Wang, Y. Pan
- Characterization of a novel impurity in bulk drug eprosartan by ESI/MSn and NMR
- J. Pharm. Biomed. Anal., 51 (2010), pp. 778–783
- [SD-008]
- [211]
- A. Mohan, S. Shanmugavel, A. Goyal, B.R. Venkataraman, D. Saravanan
- Identification, isolation, and characterization of five potential degradation impurities in candesartan cilexetil tablets
- Chromatographia, 69 (2009), pp. 1211–1220
- [SD-008]
- [212]
- S. Provera, L. Rovatti, L. Turco, S. Mozzo, A. Spezzaferri, S. Bacchi, A. Ribecai, S. Guelfi, A. Mingardi, C. Marchioro, D. Papini
- A multi-technique approach using LC–NMR, LC–MS, semi-preparative HPLC, HR–NMR and HR–MS for the isolation and characterization of low-level unknown impurities in GW876008, a novel corticotrophin-release factor 1 antagonist
- J. Pharm. Biomed. Anal., 53 (2010), pp. 517–525
- [SD-008]
- [213]
- C. Sitaram, R. Rupakula, B.N. Reddy
- Determination and characterization of degradation products of anastrozole by LC–MS/MS and NMR spectroscopy
- J. Pharm. Biomed. Anal., 56 (2011), pp. 962–968
- [SD-008]
- [214]
- W. Feng, H. Liu, G. Chen, R. Malchow, F. Bennett, E. Lin, B. Pramanik, T.M. Chan
- Structural characterization of the oxidative degradation products of an anti-fungal agent SCH 56592 by LC–NMR and LC–MS
- J. Pharm. Biomed. Anal., 25 (2001), pp. 545–557
- [SD-008]
- [215]
- A. Awasthi, M. Razzak, R. Al-Kassas, D.R. Greenwood, J.H. Sanjay Garg
- Isolation and characterization of degradation products of moxidectin using LC, LTQ FT-MS, H/D exchange and NMR
- Anal. Bioanal. Chem., 404 (2012), pp. 2203–2222
- [SD-008]
- [216]
- J. Wang, W. Li, C.-G. Li, Y.-Z. Hu
- Photodegradation of fleroxacin injection: different products with different concentration levels
- AAPS PharmSciTech, 12 (2011), pp. 872–878
- [SD-008]
- [217]
- A. Kugimiya, H. Naoki, N. Hamanaka, E. Nishimura
- Photo-degradation products of pramipexole
- Bioorg. Med. Chem. Lett., 22 (2012), pp. 2951–2953
- [SD-008]
- [218]
- R.P. Shah, S. Singh
- Identification and characterization of a photolytic degradation product of telmisartan using LC-MS/TOF, LC-MSn, LC–NMR and on-line H/D exchange mass studies
- J. Pharm. Biomed. Anal., 53 (2010), pp. 755–761
- [SD-008]
- [219]
- R.P. Shah, A. Sahu, S. Singh
- Identification and characterization of degradation products of irbesartan using LC–MS/TOF, MSn, on-line H/D exchange and LC–NMR
- J. Pharm. Biomed. Anal., 51 (2010), pp. 1037–1046
- [SD-008]
- [220]
- A.K. Gajjar, V.D. Shah
- Isolation and structure elucidation of major alkaline degradant of ezetimibe
- J. Pharm. Biomed. Anal., 55 (2011), pp. 225–229
- [SD-008]
- [221]
- C.R. Barhate, K. Mohanraj
- What is the degradation product of ezetimibe?
- J. Pharm. Biomed. Anal., 55 (2011), pp. 1237–1238
- Article
|
PDF (121 K)
|
View Record in Scopus
|
Citing articles (5) - [222]
- Z. Sánta, J. Koti, K. Szőke, K. Vukics, C. Szántay Jr.
- Structure of the major degradant of ezetimibe
- J. Pharm. Biomed. Anal., 58 (2012), pp. 125–129
- Article
|
PDF (176 K)
|
View Record in Scopus
|
Citing articles (4) - [223]
- G.Y.S.K. Swamy, K. Ravikumar, L.K. Wadhwa, R. Saxena, S. Singh
- (2R* 3R*,6S*)-N,6-Bis(4-fluorophenyl)-2-(4-hydroxyphenyl)-3,4,5,6-tetrahydro-2H-pyran-3-carboxamide
- Acta Crystallogr. E, 61 (2005), pp. o3608–o3610
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (9) - [224]
- K. Filip, K. Bankowski, K. Sidoryk, J. Zagrodzka, M. Łaszcz, K. Trzcinska, A. Szyprowska, P. Cmoch, W. Maruszak
- Physicochemical characterization of ezetimibe and its impurities
- J. Mol. Struct., 991 (2011), pp. 162–170
- Article
|
PDF (1463 K)
|
View Record in Scopus
|
Citing articles (9) - [225]
- A. Mendez, P. Chagastelles, E. Palma, N. Nardi, E. Schapoval
- Thermal and alkaline stability of meropenem: degradation products and cytotoxicity
- Int. J. Pharm., 350 (2008), pp. 95–102
- Article
|
PDF (407 K)
|
View Record in Scopus
|
Citing articles (27) - [226]
- R. Nageswara Rao, A. Narasa Raju, R. Narsimha
- Isolation and characterization of process related impurities and degradation products of bicalutamide and development of RP–HPLC method for impurity profile study
- J. Pharm. Biomed. Anal., 46 (2008), pp. 505–519
- Article
|
PDF (1514 K)
|
View Record in Scopus
|
Citing articles (19) - [227]
- R.N. Rao, Ch., G. Naidu, K.G. Prasad, B. Santhakumar, S. Saida
- Development and validation of a stability indicating assay of doxofylline by RP–HPLC: ESI–MS/MS, 1H and 13C NMR spectroscopic characterization of degradation products and process related impurities
- J. Pharm. Biomed. Anal., 78–79 (2013), pp. 92–99
- Article
|
PDF (965 K)
|
View Record in Scopus
|
Citing articles (4) - [228]
- A.J. Lin, L.Q. Li, D.L. Klayman, C.F. George, J.L. Flippen-Anderson
- Antimalarial activity of new water-soluble dihydroartemisinin derivatives. 3. Aromatic amine analogs
- J. Med. Chem., 33 (1990), pp. 2610–2614
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (56) - [229]
- C. Charrier, G. Bertho, O. Petigny, P. Moneton, R. Azerad
- A new derivative detected in accelerated ageing of artesunate-amodiaquine fixed dose combination tablets
- J. Pharm. Biomed. Anal., 81–82 (2013), pp. 20–26
- Article
|
PDF (977 K)
|
View Record in Scopus
|
Citing articles (1) - [230]
- M. Xia, T.-J. Hang, F. Zhang, X.-M. Li, X.-Y. Xu
- The stability of biapenem and structural identification of impurities in aqueous solution
- J. Pharm. Biomed. Anal., 49 (2009), pp. 937–944
- Article
|
PDF (993 K)
|
View Record in Scopus
|
Citing articles (21) - [231]
- Z. Béni, V. Háda, E. Varga, S. Mahó, A. Aranyi, C. Szántay Jr.
- New oxidative decomposition mechanism of estradiol through the structural characterization of a minute impurity and its degradants
- J. Pharm. Biomed. Anal., 78–79 (2013), pp. 183–189
- Article
|
PDF (377 K)
|
View Record in Scopus
|
Citing articles (2) - [232]
- T. Murakami, H. Konno, N. Fukutsu, M. Onodera, T. Kawasaki, F. Kusu
- Identification of a degradation product in stressed tablets of olmesartan medoxomil by the complementary use of HPLC hyphenated techniques
- J. Pharm. Biomed. Anal., 47 (2008), pp. 553–559
- Article
|
PDF (821 K)
|
View Record in Scopus
|
Citing articles (35) - [233]
- E. Del Grosso, S. Aprile, G. Grosa
- Forced degradation study of thiocolchicoside: characterization of its degradation products
- J. Pharm. Biomed. Anal., 61 (2012), pp. 215–223
- [234]
- R. Azerad
- Microbial transformations of artemisinin and artemisinin derivatives: an example of the microbial generation of molecular diversity
- Curr. Bioact. Compd., 8 (2012), pp. 151–158
- Full Text via CrossRef
- [235]
- W.-M. Wu, Y. Wu, Y.-L. Wu, Z.-J. Yao, C.-M. Zhou, Y. Li, F. Shan
- Unified mechanistic framework for the Fe(II)-induced cleavage of Qinghaosu and derivatives/analogues
- The first spin-trapping evidence for the previously postulated secondary C-4 radical, J. Am. Chem. Soc., 120 (1998), pp. 3316–3325
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (150) - [236]
- R.K. Haynes, W.C. Chan, C.-M. Lung, A.-C. Uhlemann, U. Eckstein, D. Taramelli, S. Parapini, D. Monti, S. Krishna
- The Fe2+-mediated decomposition, PfATP6 binding, and antimalarial activities of artemisone and other artemisinins: the unlikelihood of C-centered radicals as bioactive intermediates
- Chem. Med. Chem., 2 (2007), pp. 1480–1497
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (60) - [237]
- C.D. Sommers, E.S. Pang, H. Ghasriani, R.T. Berendt, V.L. Vilker, D.A. Keire, M.T. Boyne
- II. Analyses of marketplace tacrolimus drug product quality: bioactivity, NMR and LC–MS
- J. Pharm. Biomed. Anal., 85 (2013), pp. 108–117
- Article
|
PDF (1109 K)
|
View Record in Scopus
|
Citing articles (1) - [238]
- P. Ferraboschi, D. Colombo, M. de Mieri, P. Grisenti
- Evaluation, synthesis and characterization of tacrolimus impurities
- J. Antibiotics, 65 (2012), pp. 349–354
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (3) - [239]
- A. Subasranjan, R. Hemant
- An improved validated ultra-high pressure liquid chromatography method for separation of tacrolimus impurities and its tautomers
- Drug Test. Anal., 2 (2010), pp. 107–112
- View Record in Scopus
|
Citing articles (5) - [240]
- D.F. Mierke, P. Schmieder, P. Karuso, H. Kessler
- Conformational analysis of the cis- and trans-isomers of FK506 by NMR and molecular dynamics
- Helv. Chim. Acta, 74 (1991), pp. 1027–1047
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (21) - [241]
- B.E. Segmuller, B.L. Armstrong, R. Dunphy, A.R. Oyler
- Identification of autoxidation and photodegradation products of ethynylestradiol by on-line HPLC–NMR and HPLC–MS
- J. Pharm. Biomed. Anal., 23 (2000), pp. 927–937
- Article
|
PDF (179 K)
|
View Record in Scopus
|
Citing articles (37) - [242]
- M. Pendela, S. Béni, E. Haghedooren, L. Van den Bossche, B. Noszál, A. Van Schepdael, J. Hoogmartens, E. Adams
- Combined use of liquid chromatography with mass spectrometry and nuclear magnetic resonance for the identification of degradation compounds in an erythromycin formulation
- Anal. Bioanal. Chem., 402 (2012), pp. 781–790
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (7) - [243]
- C. Pan, J. Guan, M. Lin
- A multidisciplinary approach to identify a degradation product in a pharmaceutical dosage form
- J. Pharm. Biomed. Anal., 54 (2011), pp. 855–859
- Article
|
PDF (284 K)
|
View Record in Scopus
|
Citing articles (5) - [244]
- C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
- Determination of free fatty acids in pharmaceutical lipids by 1H NMR and comparison with the classical acid value
- J. Pharm. Biomed. Anal. (2013) http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.04.010
- [245]
- C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
- Determination of free fatty acids in edible oils by 1H NMR spectroscopy
- Lipid Technol., 24 (2012), pp. 279–281
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (7) - [246]
- C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
- 1H NMR spectroscopy as a new tool in the assessment of the oxidative state in edible oils
- J. Am. Oil Chem. Soc., 89 (2012), pp. 1383–1391
- View Record in Scopus
|
Citing articles (9) - [247]
- H. Böhme, K. Hartke, H. Hartke, M. Wichtl
- Kommentar zum Europäischen Arzneibuch, Vol. 41, Aktualisierungslieferung
- Wissenschaftliche Verlagsge-sellschaft, Stuttgart, Germany (2012)
- [248]
- S. Krist, G. Buchbauer, C. Klausberger
- Lexikon der pflanzlichen Öle und Fette
- Springer, Vienna, Austria (2008)
- [249]
- R.A. Seburg, J.M. Ballard, T.-L. Hwang, C.M. Sullivan
- Photosensitized degradation of losartan potassium in an extemporaneous suspension formulation
- J. Pharm. Biomed. Anal., 42 (2006), pp. 411–422
- Article
|
PDF (471 K)
|
View Record in Scopus
|
Citing articles (24) - [250]
- T.-M. Chan, B. Pramanik, R. Aslanian, V. Gullo, M. Patel, B. Cronin, C. Boyce, K. McCormick, M. Berlin, X. Zhu, A. Buevich, L. Heimark, P. Bartner, G. Chen, H. Pu, V. Hegd
- Isolation, structural determination, synthesis and quantitative determination of impurities in Intron-A, leached from a silicone tubing
- J. Pharm. Biomed. Anal., 49 (2009), pp. 327–332
- Article
|
PDF (391 K)
|
View Record in Scopus
|
Citing articles (3) - [251]
- J. Larsen, D. Staerk, C. Cornett, S.H. Hansen, J.W. Jaroszewski
- Identification of reaction products between drug substances and excipients by HPLC–SPE–NMR: ester and amide formation between citric acid and 5-aminosalicylic acid
- J. Pharm. Biomed. Anal., 49 (2009), pp. 839–842
- Article
|
PDF (269 K)
|
View Record in Scopus
|
Citing articles (15) - [252]
- P. Novak, P. Tepes, I. Fistric, I. Bratos, V. Gabelica
- The application of LC–NMR and LC-MS for the separation and rapid structure elucidation of an unknown impurity in 5-aminosalicylic acid
- J. Pharm. Biomed. Anal., 40 (2006), pp. 1268–1272
- Article
|
PDF (185 K)
|
View Record in Scopus
|
Citing articles (22) - [253]
- M. Dousa, P. Gibala, J. Havlicek, L. Placek, M. Tkadlecova, J. Bricha
- Drug-excipient compatibility testing–Identification and characterization of degradation products of phenylephrine in several pharmaceutical formulations against the common cold
- J. Pharm. Biomed. Anal., 55 (2011), pp. 949–956
- Article
|
PDF (584 K)
|
View Record in Scopus
|
Citing articles (4) - [254]
- A. Marín, A. Espada, P. Vidal, C. Barbas
- Major degradation product identified in several pharmaceutical formulations against the common cold
- Anal. Chem., 77 (2005), pp. 471–477
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (17) - [255]
- O. Galanopoulou, S. Rozou, E. Antoniadou-Vyza
- HPLC analysis, isolation and identification of a new degradation product in carvedilol tablets
- J. Pharm. Biomed. Anal., 48 (2008), pp. 70–77
- Article
|
PDF (692 K)
|
View Record in Scopus
|
Citing articles (14) - [256]
- J. Battiste, R.A. Newmark
- Applications of 19F multidimensional NMR
- Prog. Nucl. Magn. Reson. Spectrosc., 48 (2006), pp. 1–23
- Article
|
PDF (1112 K)
|
View Record in Scopus
|
Citing articles (46) - [257]
- S. Trefi, V. Gilard, M. Malet-Martino, R. Martino
- Generic ciprofloxacin tablets contain the stated amount of drug and different impurity profiles: a19F, 1H and DOSY NMR analysis
- J. Pharm. Biomed. Anal., 44 (2007), pp. 743–754
- Article
|
PDF (689 K)
|
View Record in Scopus
|
Citing articles (13) - [258]
- S. Trefi, V. Gilard, S. Balayssac, M. Malet-Martino, R. Martino
- Quality assessment of fluoxetine and fluvoxamine pharmaceutical formulations purchased in different countries or via the Internet by 19F and 2D DOSY 1H NMR
- J. Pharm. Biomed. Anal., 46 (2008), pp. 707–722
- Article
|
|
Citing articles (20) - [259]
- V.G. Dongre, P.D. Ghugare, P.P. Karmusea, S.R. Soudagar, N. Panda, A. Kumar
- Isolation and structural identification of an impurity in fluconazole bulk drug substance
- J. Pharm. Biomed. Anal., 45 (2007), pp. 422–429
- Article
|
PDF (909 K)
|
View Record in Scopus
|
Citing articles (9) - [260]
- B. Marciniec, K. Dettlaff, E. Jaroszkiewicz, J. Bafeltowska
- Radiochemical stability of fluconazole in the solid state
- J. Pharm. Biomed. Anal., 43 (2007), pp. 1876–1880
- Article
|
PDF (471 K)
|
View Record in Scopus
|
Citing articles (13) - [261]
- L. Wu, T.Y. Hong, F.G. Vogt
- Structural analysis of photo-degradation in thiazole-containing compounds by LC–MS/MS and NMR
- J. Pharm. Biomed. Anal., 44 (2007), pp. 763–772
- Article
|
PDF (644 K)
|
View Record in Scopus
|
Citing articles (12) - [262]
- J. Brusa, M. Urbanova, I. Sedenkova, H. Brusova
- New perspectives of 19F MAS NMR in the characterization of amorphous forms of atorvastatin in dosage formulations
- Int. J. Pharm., 409 (2011), pp. 62–74
- [263]
- M. Nisha, M. Ismail, R. Ismail, F. Duncan, L. Maili, K.N. Jeremy, C.L. John
- Impurity profiling in bulk pharmaceutical batches using 19F NMR spectroscopy and distinction between monomeric and dimeric impurities by NMR-based diffusion measurements
- J. Pharm. Biomed. Anal., 19 (1999), pp. 511–517
- [264]
- J.L. Battiste, N. Jing, R.A. Newmark
- 2D 19F–19F NOESY for the assignment of NMR spectra of fluorochemicals
- J. Fluorine Chem., 125 (2004), pp. 1331–1337
- Article
|
PDF (321 K)
|
View Record in Scopus
|
Citing articles (26) - [265]
- W. Zhong, B. Hilton, G. Martin, L. Wang, S.H. Yip
- Identification of a unique cationic impurity in Preladenant™ using accurate MS and NMR
- J. Heterocycl. Chem., 50 (2013), pp. 281–287
- View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (1) - [266]
- R.M. Bianchini, P.M. Castellano, T.S. Kaufman
- Characterization of two new potential impurities of valsartan obtained under photodegradation stress condition
- J. Pharm. Biomed. Anal., 56 (2011), pp. 16–22
Abstract
Current standards and regulations demand the pharmaceutical industry not only to produce highly pure drug substances, but to achieve a thorough understanding of the impurities accompanying their manufactured drug substances and products. These challenges have become important goals of process chemistry and have steadily stimulated the search of impurities after accelerated or forced degradation procedures.
As a result, impurity profiling is one of the most attractive, active and relevant fields of modern pharmaceutical analysis. This activity includes the identification, structural elucidation and quantitative determination of impurities and degradation products in bulk drugs and their pharmaceutical formulations.
Nuclear magnetic resonance (NMR) spectroscopy has evolved into an irreplaceable approach for pharmaceutical quality assessment, currently playing a critical role in unequivocal structure identification as well as structural confirmation (qualitative detection), enabling the understanding of the underlying mechanisms of the formation of process and/or degradation impurities.
NMR is able to provide qualitative information without the need of standards of the unknown compounds and multiple components can be quantified in a complex sample without previous separation. When coupled to separative techniques, the resulting hyphenated methodologies enhance the analytical power of this spectroscopy to previously unknown levels. As a result, and by enabling the implementation of rational decisions regarding the identity and level of impurities, NMR contributes to the goal of making better and safer medicines.
Herein are discussed the applications of NMR spectroscopy and its hyphenated derivate techniques to the study of a wide range pharmaceutical impurities. Details on the advantages and disadvantages of the methodology and well as specific challenges with regards to the different analytical problems are also presented.
Graphical abstract
Full-size image (35 K)
Figure options
Keywords
NMR spectroscopy;
Pharmaceutical impurities;
LC–NMR;
Structural elucidation;
Qualitative and quantitative analysis
1. Introduction
1.1. Pharmaceutical impurities and approaches to their modern quality control
The quality assurance and quality control of active pharmaceutical ingredients (APIs) and excipients are major issues in pharmaceutical analysis, which intend to prevent damages to the patients. Quality control methods are regulated in the pharmacopeias and other documents, which are continuously revised in order to keep updated drugs, excipients, and also their methods for analysis. Furthermore, internationally agreed recommendations, such as those issued by the International Conference on Harmonization (ICH) are steadily moving pharmaceutical analysis beyond compendial requirements, with the advantage of the most modern analytical technologies [1]. Pharmaceutical impurities are the unwanted chemicals that remain with the API, develop during formulation, or upon degradation of both API and drug products. Their presence, even in small amounts, might influence the efficacy and safety of the pharmaceutical products; therefore, drug purity has always been associated to drug quality.
The evolution of chemical knowledge and the advent of increasingly sensitive and selective analytical methods have been continuously stimulating the interest in the determination of drug purity and the impurities themselves in both, natural and synthetic products. This is in line with the policy of the pharmaceutical industry, which has always demanded that the API should be as pure as possible. The number of recently published books [2], book chapters [3], [4], [5] and [6] and papers on the subject demonstrate the increasing importance of pharmaceutical impurities [7], [8], [9], [10] and [11] and impurity profiling [12], [13], [14], [15] and [16]. These and other [17], [18] and [19] publications also reflect the continued interest of the regulatory authorities, industry and scientists in these areas.
The ICH Q3A guideline states that impurity profile is “a description of the identified and unidentified impurities, present in a new drug substance” [20]. According to the guide, the impurities are classified into organic, inorganic and organic volatile impurities. The organic impurities can arise from the manufacturing process and/or be formed during storage of the drug substances. They include starting materials, by-products, synthetic intermediates, degradation products, reagents, ligands and catalysts. As specifically indicated, the guide does not cover enantiomeric impurities and polymorphic forms.
The ICH Q3B guideline addresses impurities in new drug products, classified as degradation products of the drug substance or reaction (interaction) products of the drug substance with an excipient and/or immediate container closure system [21].
In addition, in current pharmaceutical development processes, impurities in the API have identification and qualification thresholds of 0.10 and 0.15% respectively, for doses of the API up to 2 g/day [20] and many recent publications reflect interest in compounds ranging from 0.01 to 0.1%.
Reliable assessment of drug purity in accordance with these stringent standards requires the use of state of the art validated analytical methods, increasingly sensitive detections, and mainly an analyst prepared to critically interpret the results.
The available tools for impurity profiling have already been reviewed [22]. Reversed-phase liquid chromatography (RP–LC) is still the primary method for analyte separation toward impurity profiling of drugs. However, an increasing number of examples show the usefulness of both nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) as additional tools for the detection and quantitation of difficult to solve impurity challenges.
When employed as stand-alone techniques, NMR and MS offer a high degree of orthogonality and complementarity [23]. In particular, NMR spectroscopy is relevant because it can simultaneously and selectively detect multiple components in a sample, even without their physical separation.
Mass spectrometric studies can provide structure-rich information of organic compounds, even those found at trace impurity levels, with small amounts of sample. When hyphenated with an LC separation, and when electro-spray (ESI) or atmospheric-pressure chemical ionization (APCI) are used, MS provides very important information about the molecular weights (especially when implemented in high-resolution mode) of the chromatographed compounds. It can also provide the structure (through the fragmentation pattern) of the analytes, especially when some prior information on the molecular skeleton is available and if only minor changes are present in the impurity with respect to the drug substance [24].
Therefore LC–ESI–MS and LC–ESI–MS/MS are currently well-established analytical tools in the rapid identification and characterization of each component in sample mixtures. However, the resulting picture is still often limited with respect to details of molecular structure. Mass spectral methods are rarely capable of de-novo structure determination with full certainty; furthermore, they do not always provide unambiguous structural information and may not be able to distinguish between isomers [25].
In these cases, the next and often final stage is to resort to NMR spectroscopy (usually off-line, but in some cases on-line), in order to access the required information. Occasionally, single crystal X-ray diffractometry and/or synthesis of the proposed structures have been used as final proofs of the impurities’ structure.
Furthermore, NMR is at the center of the new complete molecular confidence concept, which tends to exploit its synergy with LC–MS in order to obtain an estimation of the purity, concentration, and identity of chemical compounds [26].
1.2. NMR spectroscopy
1.2.1. NMR spectroscopy as an analytical tool: Advantages and disadvantages
NMR spectroscopy, an information rich analytical tool, is capable to deliver the most complete molecular structural information, including atomic connectivity, spatial geometry, conformation, and stereochemistry.1H is the most simple and sensitive, and therefore most frequently detected nucleus. Many more isotopes of elements in the periodic table (11B, 13C, 15N, 17O, 19F, 31P, etc.) are also accessible by NMR. In 1H NMR spectroscopy, 1H chemical shifts and 1H–1H couplings are selectively detected, which relate to the chemical environment of the observed nucleus. The spectral signature of each compound is unique, turning the method highly specific for chemical structures and ideally suited for structural elucidation and confirmation.
Since, the intensity of any given NMR signal depends only on the total amount of the detected nuclei in the sample volume (primary ratio rule), quantitative NMR spectroscopy (qNMR) can be used either as an absolute “standard-free” method or as a relative method [27].
The physical principles of the NMR phenomenon and the relevant spectroscopic parameters, such as chemical shift, signal multiplicity, spin–spin coupling constant (J), and the integral intensity, as well as the wide array of modern NMR experiments [28] and [29] are detailed in a number of useful books [30], [31],[32] and [33]. A short list of the most common NMR experiments and the structural information they are able to provide is found in Table 1.
Table 1.Summary of the most relevant 1H and 13C NMR experiments for studying small organic molecules.
NMR experiment Acronyma Information provided
1H NMR 1D-1H (1H) Number and types of protons.
Nuclear Overhausser effect 1D-NOE (NOE) Through space H–H vicinity.
Diffusion NMR DOSY Molecular size; host–guest interactions.
13C NMR 1D-13C (13C) Types of carbons.
Special 1D 13C NMR APT
DEPT Type of carbons (CH, CH2, CH3), according to the number of protons attached to them.
2D Homonuclear (1H–1H) correlation COSY Through bond H–H correlations (coupled protons).
TOCSY Through bond long range correlations (coupled spin network).
NOESY Through space H–H correlations.
ROESY Long range, through space H–H correlations.
2D Heteronuclear (1H–13C) correlation HMBC Short range (1J) H–C correlations.
HSQC Long range (usually 2J and 3J) H–C correlations.
a
For a list with the meaning of the acronyms used in this table and throughout the text, see Appendix A, at the end of the manuscript.
Table options
NMR spectroscopy has several advantages; it is a robust and quasi-universal detector, has a non-destructive nature, allows easy sample preparation, and simple method development. Although NMR spectroscopy is used in drug impurity profiling mainly after off-line or on-line separation of the impurities, this technique can be a useful tool even without full analyte separation. Spectra are highly specific, this spectroscopy has the ability to provide global information about the sample in a single analysis and it offers multiple calibration options.
Therefore, 1H NMR can be advantageously used as an alternative to conventional high-performance liquid chromatography (HPLC) when availability of impurity standards is not possible, such as during early stages of drug development [34]. This is documented by a steady increase in the volume of reports that employ NMR spectroscopy to solve this problem, but also by the remarkable interest of drug manufacturers in this technique. Several reviews have been published on the application of NMR in pharmaceutical analysis[35] and [36].
Since 1H NMR is sensitive to compounds bearing protons, it can be regarded as a quasi-universal detector, especially for low molecular weight organic molecules. Protons are detected with the same sensitivity, regardless of their chemical environment; thus, the requirement of determining response factors is avoided.
Therefore, NMR ranks among the most informative methods, being a key analytical tool for identification, authentication, detailed structural analysis and elucidation of organic compounds (including stereochemistry and even dynamic effects), and quantitation of unknown natural and synthetic compounds in their mixtures in a single run, provided their signals are well separated [37].
On the other side, the two main disadvantages of NMR are its high equipment cost and its comparatively low sensitivity. When related to MS [Limit of detection (LOD) in the picomolar (pM) range], NMR has lower sensitivity, with LOD values in the low mM range. However, the detection sensitivity of NMR spectroscopy may be enhanced by increasing the number of scans, the concentration of the sample solution and by employing higher magnetic field strengths or modern probes. Accordingly, recent improvements in NMR instrumentation and technology, such as shielded and increasingly stronger high-field magnets [38], cryogenic probes [39] and [40], operation with large sample volumes (several ml), solvent suppression techniques, advanced data processing, versatile pulse sequences and polarization transfer techniques[41], have increased the sensitivity up to the nanomolar range. Other improvements include small OD tubes, Shigemi tubes and microcoil technology.
These characteristics have turned NMR spectroscopy into a powerful tool that can be used in pharmaceutical analysis [42] for the quality control of APIs and excipients for identification, impurity assessment, including adulterations [43] and [44] and assay purposes. Examination of the most recognized Pharmacopeias and an inspection of the recently summarized major compendial applications of NMR spectroscopy in current pharmaceutical analysis [45], reflect an increasingly important participation of this technique in the quality control monographs.
1.2.2. NMR spectroscopy and qualitative pharmaceutical analysis
The power of NMR for structural elucidation is well established; therefore, one of the major applications of NMR experiments is the ‘de novo’ structural elucidation of small organic molecules. The latter is often considered an almost mechanical process, which can be solved mainly with the aid of NMR and/or MS methodologies, especially when the amount of sample is enough to run the powerful two-dimensional NMR (2D-NMR) experiments.
In practice, however, in the case of impurities this represents a rather intellectually and technically challenging problem, because their amounts are often scarce [46], [47], [48] and [49] and their structure is not always related directly to the API. Structural elucidation of trace impurities without separation poses additional demands, requiring minimum signal overlapping between structurally useful spectral signals of the impurity and the resonances of the main component.
However, the information provided by NMR is of such magnitude that the technique can be even used to determine the origin (production plant) of a drug based on its spectral profile [44].
1.2.3. NMR spectroscopy and quantitative pharmaceutical analysis: qNMR
The basic principles of quantitative NMR (qNMR) have been discussed elsewhere [50]. The main advantage of NMR spectroscopy is the linear relationship between the integrated intensities of the resonances from separate components in the spectrum and their molar contents in the sample [27]. The quantitative applications (1H qNMR, qHNMR) are based on the nearly equal response of protons, regardless of their chemical shifts or coupling to other nuclei.
Absolute quantification is possible by relating the peak area of interest in the sample to a signal from an appropriate internal standard, without the need for a reference standard of the same chemical structure as the sample [51] and [52]. When NIST (National Institute of Standards and Technology) standards are used, traceability to the International System of Units (SI) can be established. Relative quantification can be carried out by relating the intensity of the peak of interest to one in the main compound.
In addition, NMR spectroscopy can be considered a relative primary analytical method, because it can be described completely by mathematical equations from which a full uncertainty budget may be derived[53] and [54]. A validation scheme for qNMR methodologies has been proposed [55].
The technique offers a valuable, un-biased and near-universal approach for the analysis of complex mixtures and the simultaneous quantitation of their components, with minimum sample preparation and without the need of a previous separation. Therefore, qHNMR spectroscopy is highly regarded for use in purity assessment of small organic molecules, complementing the identification of potential impurities [56],[57] and [58]. Currently, sensitivity and accuracy of the quantitative results are suitable for controlling low level impurities and qHNMR measurements are at least as reliable and precise as those obtained by chromatographic techniques [59].
Therefore, qNMR is increasingly gaining interest in pharmaceutical applications and has become an essential tool that may be used for establishing sample composition and components’ molar ratio, as well as for assigning content, including the determination of the level of impurities [49]. Furthermore, the capability to perform concurrent identification and quantitation of impurities turns qHNMR into a unique analytical tool. However, the most challenging item is to find proper conditions and separated, pure signals, especially during impurity profiling [60].
An advantage of NMR is that quantitation can be achieved using the main sample component signals thus alleviating the need for time-consuming system suitability and standard runs prior to sample analysis [61]. This strong point was stressed by Deubner and Holzgrabe during their analysis of complex aminoglycoside antibiotics by micellar electrokinetic chromatography, RP–LC and NMR. In that study the authors described the difficulty of evaluating the impurities in gentamicin sulfate which contains no chromophore and has many structurally similar aminoglycoside impurities.
1.2.4. Hyphenated NMR techniques
LC–NMR is one of the most powerful techniques for the characterization of complex mixtures, since it combines a very efficient separation technique and the most important structure elucidation method. However, peak isolation can be also performed by liquid or gas chromatography (GC), as well as by other methods such as capillary electrophoresis (CE). These methods are very different, particularly with respect to sensitivity.
Only recently has LC–NMR coupling become efficient enough and hyphenation allowed the long-expected gain of maximal on-line information about individual constituents of a mixture, with due speed and efficiency. Details of the instrumentation and potential are given in many specific books [62], [63],[64] and [65]; its general applications in pharmaceutical analysis have also been detailed [66].
Analogous hyphenations [67] such as GC–NMR [68] and [69], CZE–NMR [70], [71] and [72], CEC–NMR[73] and [74] and CE–NMR [75], [76], [77] and [78] have also been explored. However, in the on-line mode none of them have been used to solve problems in the area of pharmaceutical impurities and currently this application seems still unfeasible, mainly due to sensitivity problems. To date, CE–NMR is not a well-established method; however, development of smaller volume NMR probes and continuous enhancements of NMR sensitivity will probably transform CE–NMR in the near future into a valuable technique for biopharmaceutical analysis. On the other hand, despite its years of evolution and recent advances, GC–NMR is still in its infancy [79]; even more, its sensitivity, interfacing and other instrumental details require strong improvements.
In its different modes (on-flow, stopped flow and loop-storage techniques) HPLC–NMR may allow unambiguous on-line identification of even very complex structures. An integrated LC–NMR and LC–MS approach toward structural elucidation of impurities without their isolation has been informed [80]. However, very often it is learned that subsequent to having spent a considerable time on separation method development, the amount of structural information collected is limited, because it relates to the amount of time the sample remains in the flow cell. Fraction collection is of help, but attempts to isolate an impurity for off-line structure investigation does not always yield pure substances; instead, the resulting mixtures many times are not easily amenable to analysis, or the quantities are so small that they hinder running 2D NMR experiments.
A recent extension capable of providing NMR data of higher quality includes the insertion of a solid-phase extraction (SPE) unit between the HPLC column and the NMR probe [81], which was also employed for detecting adulterants in an herbal medicine [82]. This HPLC–SPE–NMR setup, where lipophilic stationary phases (silica-C18, styrene-divinylbenzene) are commonly utilized as SPE materials, allows the on-line trapping of the impurity peaks while removing the HPLC eluent.
Use of NMR solvents of high elution power (CD3CN, CD3OD) allows matching the peak volume to the active volume of the NMR flow cell, when the impurities are automatically back-flushed, submitting them to the NMR probe; this results in an overall increase of the sensitivity of the measurement due to analyte accumulation by multiple SPE collections. The overall process requires careful optimization of the SPE trapping and elution conditions, and it must also take into account that use of lipophilic stationary phases may result in loss of the more polar impurities, that cannot be trapped by the SPE stationary phase.
The problem of isolating the impurity for conventional NMR analysis (∼1 mg) can be greatly alleviated by using cryogenically cooled probes, which offer an approximately fourfold increase in S/N, reducing the amount of impurity needing to be purified. Flow and non-flow alternatives are possible, as well as combination with SPE [83]. However, in many cases this does not alleviate the burden of manual preparative-scale isolation of the impurities.
The use of LC–NMR for identification of drug impurities has been impulse during the last 20 years [84]. In this way, NMR and particularly LC–NMR analysis provide complementary information to MS for rapid and unequivocal structural determination. This feature and the appearance of accessible and more powerful spectrometers designed for routine measurements and by the simplicity of the measurements themselves explain the high and increasing interest in NMR spectroscopy on the part of pharmaceutical analysts [46].
2. NMR-assisted Identification/structure elucidation and quantitation of related impurities
According to the ICH Q3A Guideline [20], a “potential impurity is an impurity that theoretically can arise during manufacture or storage”. It has been pointed out that NMR spectrometry is finding increasing application for the identification of unknown organic impurities in pharmaceutical preparations [85]. Impurity profiling is a typical team effort which involves many types of analytical chemists, including UV, IR and NMR spectroscopists, mass spectrometrists, TLC, HPLC and GC chromatographers, as well as experts in hyphenated and several other techniques. When NMR spectroscopy is also necessary for the structure elucidation [86], somewhat larger sample size is necessary. The use of impurity-enriched mother liquors or crude reaction products can overcome problems associated with sensitivity and may be adopted as a general strategy for their identification [87]. These samples are usually obtained by semi-preparative HPLC [88] or by column chromatography.
2.1. Process-related impurities
2.1.1. Impurities resulting from isolation in naturally occurring APIs
Echinochrome is a quinoid pigment isolated from some sea-occurring invertebrates, which is used in ophthalmology and for the treatment of acute myocardial infarction and ischemic heart disorder. Seven new impurities were isolated from the bulk drug and its formulation and their structures were elucidated by NMR in combination with other spectroscopies. The physicochemical properties of the impurities are close to those of echinochrome. For this reason, they can be present in echinochrome bulk drug after technological operations of isolation and purification. Some of these are also natural products [89].
NMR spectroscopy was employed to identify and quantitate impurities in dihydroquercetin (DHQ) [90]. Quercetin, related flavonoids (naringenin, kaempferol, dihydrokaempferol) and other species, resulting from plant raw material extraction, and impurities resulting from the extraction process, such as acetone, could be identified and quantitated from the proton spectra.
The qualitative and quantitative composition of the impurities is unique for the raw material and the DHQ production method. Therefore, it was proposed that 1H NMR spectra are good fingerprints for determining the origin and preparation method of drugs based on DHQ.
Salidroside has been widely used in Chinese traditional medicine for its antioxidant and antiapoptotic properties and is a drug candidate for the treatment of cardiovascular and cerebrovascular diseases. Three impurities with related chemical structures were found during the analysis of the bulk drug and characterized by various 1D and 2D NMR techniques and other spectroscopies [91]. A pathway for their biosynthesis was also proposed, as shown in Scheme 1.
Full-size image (26 K)
Scheme 1.
Biosynthetic pathway of salidroside and its impurities.
Figure options
Sodium tanshinone IIA sulfonate (STS) is a water-soluble derivative of tanshinone IIA, an important lipophilic component isolated from the roots of Salvia miltiorrhiza. As an injection, STS is widely and successfully used in China for treating cardiovascular diseases. In the analysis of STS bulk drug, eight related impurities were observed and isolated by column chromatography, which structure was established employing a combined NMR and LC–ESI/MS approach [92]. NMR experiments (1H, 13C, HMBC, HSQC, COSY) were crucial for unequivocally establishing structural features, where MS was not useful, allowing to propose a possible mechanism for the formation of the impurities. Some of the impurities found were known related compounds [93].
NMR spectroscopy was also employed for the structural elucidation of previously unknown impurities coming from new isolation processes. The traditional procedures for isolating vincristine and vinblastine from Catharanthus roseus are relatively inefficient. Recent efforts have resulted in a more effective manufacturing process at the expense of the appearance of new impurities. These and other known impurities were unambiguously identified by NMR spectroscopy, combined with MS techniques[94] and [95], after preparative chromatographic isolation of impurities. An 800 MHz NMR spectrometer equipped with a 1H{13C/15N} Triple Resonance 13C Enhanced Salt Tolerant Cold Probe was employed, where 2D experiments could be run overnight with 10 mg samples. 1H–1H, direct 1H–13C, long-range1H–13C scalar and dipolar spin–spin connectivities were established from a combination of 1D (1H, 13C) and 2D (gHSQCAD, zTOCSY, gHMBCAD, NOESY and ROESY) NMR experiments. A DBPPSTE DOSY diffusion experiment was also performed.
Collection of the required spectral data was made more difficult by the relatively large size of the molecules, which yielded crowded spectra, turning laborious the extraction of spin–spin connectivities. In addition, these bisindoles exhibit solvent-, field-, and temperature-dependency of the sign and size of the 1H–1H NOEs, serious solvent-dependent line-broadening that affects different signals differently in any given solvent, or line splitting due to slow N-formyl rotamerism in vincristine analogs. These difficulties were overcome by running the experiments in two or three different solvents and using the whole set of data in a complementary fashion in order to extract all the required information.
Applying the mass spectrometric shift technique to HPLC–ESI–MS/MS analysis, four impurities were identified in the unique semi-synthetic vinca alkaloid vinorelbine bitartrate. These were isolated by prep-HPLC and their structures were further confirmed by means of 1D and 2D (HMBC, HMQC, COSY) NMR spectra [96].
Most amino acids lack proper chromophors for UV-detection; therefore, the determination of low levels of impurities is analytically challenging [97] and [98]. Amino acids are produced by different manufacturing processes, each one conveying to the product a different impurity profile, with fermentation resulting in the most complex array of impurities [99]. NMR was employed as an orthogonal technique to HPLC for quantitative analysis of potential impurities of alanine. Organic acids such as malic, fumaric, aspartic and glutamic, were assessed by 1H NMR spectroscopy with 400 and 600 MHz spectrometers using 128 and 16 scans, respectively [35] and [98]. In addition, L-Isoleucine and L-leucine present as impurities in L-valine are responsible for some of the process impurities found in lopinavir [100]. The latter were characterized by FT–IR, MS and 1H NMR techniques.
During the 2007–2008 heparin crisis it was detected the presence of oversulfated chondroitin sulfate (OSCS), the adulterant of the API which caused many deaths [101]. NMR spectroscopy has been in use for fractionated medium-sized heparins more than a decade before this case. This event, however, where NMR spectroscopy played a decisive role in the structural determination of the contaminant, spurred intense research activity in this area, which resulted in a series of tests orthogonally aiming to achieve identification, measurement of bioactivity and detection of process impurities or contaminants in these drug products [102]. As a result, the current enoxaparin USP monograph contains a 13C NMR spectroscopy test and the heparin sodium monograph includes a 1H NMR identification test [103].
1H NMR spectroscopy is very sensitive to minor structural variations, allowing easy identification of the repeating pattern of the disaccharide units of heparin. Therefore, not surprisingly NMR spectroscopy proved to be a powerful tool for quali- and quantitative analysis of compounds related to the extraction and purification processes of heparin, including dermatan sulfate, chondroitin sulfate A, galactosamine and 3-O-acetylated uronic acid derived impurities [104], [105], [106] and [107].
In these cases, major advantages of NMR analysis are its unique ability to unambiguously determine sulfation locations and the stereochemistry of the anomeric bonds between the saccharide subunits. The most useful experiments for spectral assignment are COSY and TOCSY (homonuclear correlation), NOESY and ROESY (nuclear Overhauser effect), HSQC and HMQC (heteronuclear single or multiple quantum coherence spectroscopy). Fortunately, some of the characteristic resonances of these contaminants lie outside the heparin regions, easing their detection; chemometrics analysis of the data resulted in more powerful methods [108], [109], [110] and [111].
Besides the use of NMR spectroscopy for impurity detection and characterization, scattered articles have reported the use of qHNMR as a way to determine the purity of isolated plant metabolites, along with the identity of their impurities [112], [113] and [114].
2.1.2. Impurities in synthetic active pharmaceutical ingredients
APIs are usually prepared through multistep organic synthesis. Process-related impurities can be formed at any step and can ultimately appear in the final bulk drug, especially during scale-up of early-stage drug candidates. These low-level impurities formed by side reactions are always problematic, since they may affect the API through an unwanted pharmacologic activity or just merely from the cosmetic point of view, when colored by-products are formed. Therefore, it is always desirable to investigate the structures of by-products not only in the reactants but also in intermediates and final products. Elucidation of the structures of these process impurities is an important step in refining the process chemistry in order to develop robust routes for manufacturing APIs.
Different techniques, including LC coupled to ion-trap and time of flight mass spectrometry (IT/MS, LC–TOF/MS) and NMR (1H, 13C, DEPT, and HETCOR) allowed characterization of impurities in chloroquine and hydroxy chloroquine bulk drug samples [115]. Process-related impurities were also detected in ezetimibe by LC–MS/MS and elucidated by NMR. NOESY experiments were relevant in this investigation[116] and [117]. An impurity was found in phenazopyridine hydrochloride; its identification by MS–MS and 2D-NMR spectroscopy allowed to propose a mechanism for its formation [118]. Analogously, a tridesoxy impurity found in didanosine and characterized by 300 MHz NMR (1H and 13C) was proposed to result from deoxygenation during the hydrogenation stage of the preparation of the API [119].
Six process-related impurities were detected in pantoprazole sodium bulk drug substance [120] and another six impurities were found in the structurally related rabeprazole [121]. These impurities differed mainly in the degree of oxidation of both, the sulfur bridge and the pyridinic nitrogen. They were identified, synthesized and characterized using 1H, 13C and DEPT NMR experiments, aided by techniques such as HPLC, LC–MS and IR. Two impurities of tazarotene were characterized by NMR [122].
Process-related impurities found in triclabendazole were identified by 1D and 2D NMR (1H, 13C and DEPT, HSQC, HMBC) spectroscopy, concluding that one of the impurities originates in an impurity of the starting materials [123]. Glutamine as an impurity of glutamic acid generated a process related impurity in leucovorin [124]. Synthesis and spectroscopic characterization (IR, MS, 1H and 13C NMR) of the impurity provided the definitive proof for its postulated structure. The process related impurities in ceftizoxime sodium bulk drug were identified, isolated and characterized by using HPLC (analytical and preparative), LC–MS–MS, IR and NMR (1H, 13C) techniques. Spectra of the impurities and the API were compared [125].
Process-related impurities were also found in telmisartan [126], valsartan [127] and candesartan cilexetil[128], and elucidated/confirmed based on 2D-NMR and MS data. Three impurities were detected in simvastatin substance and tablets By HPLC–DAD. Their proposed structures revealed modifications of the lactone ring of the simvastatin molecule; one of them was synthesized and structurally characterized by NMR [129]. For the development of an HPLC separation, process-related impurities of pridinol mesylate were also synthesized and characterized by 1H and 13C NMR spectroscopy [130].
Classical methods were also employed for the isolation and characterization of process related impurities in eslicarbazepine [131]. A potential process related impurity of phenazopyridine HCl was isolated by preparative HPLC and structurally elucidated by MS–MS and 2D-NMR spectroscopy [118]. Analogously, four process-related impurities were found in the novel antiepileptic drug retigabine [132], which included (Scheme 2) the reduced unreacted starting material (Impurity-1), a defluorinated analog (Impurity-2), the solvent-acylated final product (Impurity-3) and the last intermediate (Impurity-4). Their structures were determined by 1D (1H, 13C, DEPT, 19F), and 2D (1H–1H COSY, HMBC, HSQC) NMR experiments.
Full-size image (40 K)
Scheme 2.
Synthetic sequence of the manufacturing process of retigabine and sources of the impurities.
Figure options
Eight related impurities of olanzapine were isolated and characterized, including a new impurity confirmed as 1-(5-methyl-thiophen-2-yl)-1H-benzimidazol-2(3H)-one by X-ray single diffraction, MS, 1H NMR, 13C NMR and HSQC [133]; a mechanism for its formation was proposed.
The structural determination [IR, MS and NMR (1H, 13C, DEPT)] of four impurities in zafirlukast allowed to propose a synthetic scheme to account for their origin [134]; analogously, four impurities were found in montelukast; after their characterization by spectroscopic analysis and the corresponding synthesis, their origin was also proposed [135].
Some examples found in recent literature point out to special precautions required during complex syntheses. The structural analysis (MS, IR, and 200 MHz 1H and 13C NMR) of an impurity isolated from lisinopril revealed it to be a result from incomplete protection of the starting material [136]. An impurity was isolated from benazepril and subjected to spectroscopic analysis using LC–MS/MS, MS, NMR (1H and 13C) and FT-IR. From its structure, it was proposed that it might have been generated due to the presence of small amounts of moisture during a key hydrogenation step [137].
Zolmitriptan-dimer, which was identified as by-product of the last step Fischer indole synthesis, was characterized as an impurity in zolmitriptan by means of LC–MS and NMR studies [138]. Two new dimers (1,2 and 2,2) were found as process impurities in rizatriptan. From the 1H and 13C NMR values it was concluded that N,N-dimethyl butanal diethyl acetal which is used as reagent in the synthesis of rizatriptan is reacting at indole 2-position of one rizatriptan molecule and at reactive positions of another rizatriptan molecule, forming a dimer with loss of ethanol [139].
Three process impurities were observed in adapalene and characterized by 1H and 13C NMR spectroscopy. Taking into account the synthetic process of the API, one of them is formed during the Friedel–Crafts step employed to prepare the first intermediate, while the others are formed during the Negishi coupling of the latter. Their presence is diagnostic of the involvement of these steps in the corresponding synthetic procedure [140].
After a systematic revision of the synthetic process, the structural elucidation and quantitative determination of seven process impurities (two of them new) of the melatonergic agonist agomelatine was carried out, employing reference standards of the impurities and with the help of NMR and FT-IR techniques [141]. This allowed the development of a new chromatographic method for analysis of the API (Scheme 3).
Full-size image (46 K)
Scheme 3.
Manufacturing process of agomelatine and origin of the potential impurities originating in side reaction and degradation.
Figure options
Relevant impurities were found in citalopram and escitalopram; their structures were elucidated by MSn(ion trap mass analyzer), accurate mass (Q-TOF mass analyzer), FT-IR and NMR (1H, 13C NMR and DEPT) data [142] and [143]; this enabled to propose a mechanism for their formation. A process impurity in a crude roflumilast preparation was isolated, characterized by NMR spectroscopy and its identity confirmed by an independent synthesis [144].
Four process and degradation impurities were found in darifenacin. Based on the structures of the impurities, proposed by LC–MSn, these were independently synthesized and used for further structural confirmation by IR, NMR and MS techniques [145].
2.1.3. Process impurities: Geometric and conformational isomers
The geometric isomer Z of endoxifen was confirmed as the active isomer through 2D NMR experiments, including HSQC and COSY [146]. NOE (ROESY) experiments were employed to establish spatial proton–proton correlations between key protons on the ethyl group and the neighbor aromatic rings and confirm the stereochemistry of each isomer ( Fig. 1).
Full-size image (18 K)
Fig. 1.
1H and 13C NMR diagnostic signals of Z– and E-endoxifen (ppm values).
Figure options
The major impurity of the drug substance was found to be the E-isomer. The previous identification of the geometric isomers of endoxifen by 1H NMR was empirically based on reported trends for the chemical shifts of the single bondOsingle bondCH2 protons in the side chain, mentioned to be upfield in the (Z)- compared to (E)-configuration for similar triarylethylene compounds [147].
The identification and characterization of a geometrical isomeric photo degradation product of eprosartan was reported, using LC–MS and LC–NMR [148].
Five impurities were isolated from a crude reaction mixture of glimepiride. These were spectroscopically characterized by NMR, FT-IR, UV and MS; among them, the cis (geometric isomer), as well as the metaand ortho (positional) isomers were found [149]. On the other hand, the 1H NMR spectrum of enalaprilat exhibits two separate (Δδ ≈ 0.2 ppm) sets of signals. They correspond to the cis and trans conformers of the drug [150], which are in equilibrium with a isomer ratio of 71.5:28.5 at 298 °C, and arise from the slow rotation around the amide bond.
2.1.4. Process impurities: Positional and functional group isomers
A positional isomer of primaquine was detected as an impurity. The proposed compound was synthesized and characterized by NMR spectroscopy [151]. Analogously, four impurities were detected in piperaquine phosphate API by a newly developed gradient RP–HPLC method. They were identified by LC–MS/MS and their structures, which relate to the manufacturing process, were confirmed by NMR and FT-IR spectroscopy, conducted using synthesized authentic compounds. A positional isomeric impurity was identified by 2D NMR spectroscopy (1H–1H COSY, HSQC, HMBC). This impurity is formed because of contamination of batches of the 4,7-dichloroquinoline precursor with 4,5-dichloroquinoline [152] and [153].
LC–MS was employed to detect an isomeric impurity in cefpodoxime proxetil, a third generation cephalosporin, and its hydrolysis product. Detailed structural information was confirmed by LC–NMR [154]. Isomeric impurities in cefdinir were confirmed in an analogous fashion [155].
Ivermectin is produced by hydrogenation of a series of avermectins. Examination of ivermectin by LC–MS data revealed four process impurities. In one of them the fragmentation pattern was identical to avermectin B2a indicating a possible isomer; this suspicion was confirmed by NMR spectroscopy [156].
The related compounds of the novel hypoglycemic drug G004 were identified. Using 1H, 13C NMR and 2D NMR analysis and MS data, it was found that one of the isolated compounds was an isomer of the drug[157]. Etimicin sulfate is a semi-synthetic aminoglycoside prepared from gentamicin C1a. Ten impurities were detected in the bulk drug by LC–ELSD and LC–ESI–MSn, many of them followed the similar fragmentation pattern as a result of differing in the position of an ethyl substituent. Six of them were isolated by column chromatography and two impurities proved to be isomers of the drug after 1H and 13C NMR analysis and comparison with literature data [158].
1H and 13C NMR spectral analysis helped elucidating a C-alkylated isomeric impurity (functional group isomer) in duloxetine hydrochloride [159] and [160], a O-alkylated naphthol ( Fig. 2). The formation of this unusual reaction product was attributed to the ambidentate nature of the nucleophile naphthol.
Full-size image (17 K)
Fig. 2.
Chemical structures and diagnostic NMR resonances (in ppm) in duloxetine and its impurity.
Figure options
2.1.5. Process impurities: Configurational isomers. Epimers and enantiomers; NMR determination of enantiomeric purity
Molecules having stereogenic centers are highly important as pharmaceutical products; some of them are natural products, but semi-synthetic and fully synthetic small organic molecules constitute an increasing proportion of these APIs in the current pharmaceutical arsenal, many of which are sold as a sole optically active enantiomer. The ICH Q6A guideline states that “where a new drug substance is predominantly one enantiomer, the opposite enantiomer is excluded from the qualification and identification thresholds given in the ICH Guidelines on Impurities in New Drug Substances and Impurities in New Drug Products because of practical difficulties in quantifying it at those levels. However, that impurity in the chiral new drug substance and the resulting new drug product(s) should otherwise be treated according to the principles established in those Guidelines” [161].
Accordingly, the quality control of chiral reagents and products requires methods capable of discriminating between enantiomers and for determining their enantiomeric purity, to resolve questions such as whether racemization has occurred during the synthesis of a chiral molecule or distinguish the enantiomeric impurity among other impurities in a sample. Typically, a 98% pure catalyst, ligand or auxiliary reagent will intrinsically afford at least 2% of the wrong enantiomer, lowering the enantiomeric excess of the final product. Therefore, since 1998 the control of impurities in chiral reagents [162], [163] and [164], involves NMR among other methodologies, is a routine task.
Currently, the method of choice for limiting enantiomeric impurities is chromatography (GC, HPLC) with chiral columns. Among the other alternatives, NMR spectroscopy is the only non-separative technique useful for the determination of the stereochemical purity of materials of pharmaceutical interest, employing for that purpose chiral derivatizing agents, lanthanide shift reagents and chiral solvating agents (CSA). CSA-based chiral NMR methods can be developed when only a single enantiomer of the analyte is available [165], provided both enantiomers of the CSA are at hand. This and other developments have converted 1H NMR into a straightforward and more time-efficient approach toward detection and quantification of enantiomers [166] and [167].
NMR spectroscopy is also well known to be one of the most powerful tools for the elucidation of the chiral recognition mechanism on a molecular level [168]. NMR signal separation in chiral environments has been studied and the corresponding mechanisms were related to those operating in separative techniques such as HPLC and CE [169]. The chiral recognition can also be observed by 13C NMR spectroscopy [170]; however, since this is a less sensitive alternative, the method is not used as frequently as 1H NMR.
The enantiomeric purity of acyl-l-carnitine was determined by 500 MHz 1H NMR employing fast diastereomeric interaction with chiral shift reagents such as chiral lanthanide-camphorato {[Eu(hfc)3], [Pr(hfc)3]} or chiral samarium-pdta shift reagents. The LOD of the determination was 0.5% for the D-enantiomer [171].
NMR is also a powerful tool for distinguish between diastereomers. Seven related impurities were detected in asperosaponin VI bulk drug and their structures were elucidated by TOF-MS, IR and NMR techniques. One of the impurities was found to be a mixture of two epimers (α- and β-glucosyl anomer derivatives)[172]. Four impurities were detected in the semisynthetic drug docetaxel. Two of them are oxazolidinones resulting from side reactions during the introduction of a Boc protecting group and acid deprotection, while the remaining pair are epimers of the API, formed by isomerization of two different hydroxyl groups[173] and [174].
Three process impurities were recently detected in the fluorine-containing steroid clocortolone pivalate by HPLC. They were isolated by semi-preparative LC and identified by MS and NMR spectroscopy. One of them was the 6β-fluor epimer of the API. Their formation was also proposed [175]. Table 2 summarizes the role of NMR spectroscopy in providing improved knowledge on process impurities of pharmaceutically relevant active ingredients.
Table 2.Process impurities found in some active pharmaceutical ingredients.
Drug Field (MHz) NMR experiments Remarks Ref.
Agomelatine 500 1H, 13C, DEPT, COSY, HMBC, HSQC Imp-1: First intermediate.
Imp-2 and Imp-3: Side reaction products of Imp-1.
Imp-4: Acetylated derivative of Imp-2.
Imp-5: Starting material.
Imp-6: Dimer of Imp-1.
Imp-7: Acetylated derivative of Imp-6. [141]
Chloroquine (CQ) Hydroxychloroquine (HCQ) 400 1H, 13C, DEPT, COSY, HETCOR (1H–13C) Imp-1 and Imp-2 (CQ-I and HCQ-I): Formed by deethylation of intermediates 5-(N,N-diethyl-ethylamino)-2-pentyl amine and 5-(N-ethyl-N-2-hydroxyethylamino)-2-pentyl amine, respectively.
Imp-3 and Imp-4 (CQ-II and HCQ-II): Quaternary ammonium products formed by chloromethylation of the amino side chain of the APIs in basic medium, with dichloromethane. [115]
Citalopram 400 1H, 13C, DEPT-135 Imp-1: The Grignard reagent performs a double addition in the first step of the synthesis. The product undergoes all the following reactions toward the API.
Identified as 1-(1,1-bis(4-fluorophenyl)-1,3-dihydro isobenzofuran-5-yl)-4-(dimethylamino) butan-1-one HBr. [142]
Clindamycin palmitate hydrochloride 300 1H, 13C, DEPT Imp-1 (Clindamycin palmitate sulphoxides α/β-isomers): Formed by oxidation at the sulfur atom.
Imp-2, Imp-4, Imp-7 Imp-9 and Imp-10 (laurate, myristate, pentadecanoate, heptadecanoate and stearate ester of Clindamicin respectively): From impurities in the starting palmitoyl chloride.
Imp-3 (Lincomycin palmitate): Lincomycin is the starting material for the synthesis of Clindamycin.
Imp-5 (7-epiclindamycin palmitate): Formed by C-7 epimerization.
Imp-6 (Clindamycin palmitate 3-isomer): Formed by esterification on C-3.
Imp-8 (Clindamycin B-palmitate): Clindamycin B present as an impurity in the starting Clindamycin.
Imp-11 and Imp-12: Starting palmitic acid and Clindamycin. [177]
Clocortolone pivalate 200, 300, 600 1H, 13C, NOESY Imp-1: 6β-Fluor epimer.
Imp-2: 4-Fluor isomer.
Imp-3: 11-Pivaloylated Δ60,07 derivative. [176]
Escitalopram 400 1H, 13C, DEPT, HMBC, HSQC, DQF-COSY Imp-1 (ESC-I): Uncyclized precursor of the API.
Imp-2 (ESC-II): Formed by chloromethylation of the amino side chain of ESC with CH2Cl2(solvent).
Imp-3 (ESC-III): Formed by chloromethylation of the amino side chain of ESC-I with CH2Cl2. [143]
Eslicarbazepine acetate 400 1H, 13C, COSY Imp-1: Due to the presence of propionic anhydride in acetic anhydride, used in the last step of the synthesis. [131]
Ezetimibe 500 1H, 13C, DEPT, COSY, HSQC, HMBC Imp-1: Formed during hydrogenation of the last intermediate (benzyl ezetimibe), by deoxygenation of the benzyl alcohol with cleavage of the β-lactam C-N bond.
Imp-2: Formed by deoxygenation of the benzyl alcohol moiety. [117]
G004 500 1H, 13C, HMBC Imp-1: 1-(4-(2-(2-bromobenzenesulphonamino) ethyl) phenylsuphonyl)-3-(trans-4-methyl cyclohexyl) urea is the 2-bromo analog. Isolated using LC. [157]
Montelukast sodium 500 1H, 13C, DEPT, 2D (unspecified experiments) Imp-1: Result of incomplete transformation of the cyano group into a carboxate during the basic hydrolysis stage.
Imp-2: Formed during the Montelukast work up stage by protonation and subsequent dehydration of the tertiary hydroxyl moiety.
Imp-3 and Imp-4: Saturated and deschloro analog of the starting material, which undergo all the sequence of reactions leading to the API. [135]
Naproxen 400 1H, DQFCOSY, gHSQC, gHMBC Imp-1: Identified as 2-(6-methoxynaphthalen-2-yl) acrylic acid, the acrylic acid of naproxen. [33]
Phenazopyridine.HCl 400 1H, COSY, NOESY Imp-1: Formed by addition of phenyl radical to phenazopyridine HCl during last step of the synthesis. Identified as 3-phenyl-5-phenylazo-pyridine-2,6-diamine. [118]
Retigabine 400 1H, 13C, 19F, COSY, HMBC, HSQC Imp-1: Produced by reduction of ethyl 2-nitro-4-aminophenylcarbamate as a vestigial intermediate.
Imp-2: Resulting from benzaldehyde being an impurity in 4-fluoro benzaldehyde.
Imp-3: Acetylation product of retigabine.
Imp-4: Vestigial intermediate of retigabine. [132]
Rizatriptan Benzoate 300 1H, 13C Imp-1: Formed during the Fischer indole reaction, rizatriptan further undergoes a dimerization reaction through electrophilic substitution, yielding 1,2 and 2,2 rizatriptan dimers. [139]
Sodium
Tanshinone IIA Sulfonate 300, 500 1H, 13C, COSY, HMQC, HMBC Imp-1 and Imp-5: Hydroxylated derivatives of the API.
Imp-2: Formed by removal of the methyl group at C-4 of Imp-4.
Imp-3 and Imp-6: Side products attributed to oxidation of C-19.
Imp-4: Formed by dehydration of Imp-1.
Imp-7: Formed by hydration of the furan ring of Imp-1.
Imp-8: Produced by oxidation and further esterification at C-19 of Imp-4. [92]
Tazarotene 300 1H, 13C Imp-5: Side reaction of a PCl3-mediated Pummerer rearrangement.
Imp-6: Side product of a Sonogashira coupling reaction. [122]
Telmisartan 300 1H, 13C, DEPT, COSY, HSQC, HMBC Imp-1: Formed during radical bromination of the starting material 4-methyl biphenyl-2-carboxylate. [126]
Valsartan 400 1H, 13C, DEPT Imp-1 and Imp-2: Synthetic intermediates.
Imp-3 and Imp-4: Products resulting from free radical desulfurization.
Imp-5: Result from reaction of 5-phenylthiovaleric acid with Imp-1 and subsequent alkaline hydrolysis. [127]
Vincristine (VCR)
Vinblastine (VLB) 800 1H, 13C, 15N, COSY, HSQC, NOESY, ROESY, HMBC, gHMBCAD, 1H–15N gHSQCAD, zTOCSY Three new impurities.
Imp-2: Cyclo-VCR.
Imp-4: [VCR]-C(16)-COOEt, from oxidization of Imp-5.
[VLB]-C(16)-COOEt: This impurity is a new natural product. [94] and [95]
Zafirlukast (ZLK) 200, 400 1H, 13C, DEPT Imp-1: Formed by trans-esterification of the cyclopentyl group of ZLK with methanol under basic conditions.
Imp-2: Formed from Imp-1 by condensation with o-toluene sulfonamide.
Imp-3 and Imp-4: Due to the presence of m/p-toluene sulfonamide impurities in o-toluene sulfonamide.
Imp-5: Synthetic intermediate. [134]
Table options
2.1.6. Genotoxic, toxic and mutagenic impurities
Genotoxic impurities (GTIs) are chemical compounds that may be mutagenic and could potentially damage DNA with an accompanying risk of cancer.
The analysis of GTIs has many special characteristics and difficulties. First of all, it can be very challenging because usually they must be controlled at levels significantly lower than 0.1% (1000 ppm). According to the current guidance, the threshold for toxicological concern in commercial products is 1.5 μg/day. Current regulations expect to control GTIs to levels in the ppm range [176]. Therefore, the analytical procedure should ideally allow detection limits in the range of 1–5 ppm.
Another special issue is solubility, since high sample concentrations are required (100 mg/ml) to enable analyses of the low-level GTIs. This poses the risk of sample precipitation, especially if the sample solution needs to be cooled in order to control degradation.
In addition, there is no single analytical procedure for analysis of all GTIs, because of their wide structural diversity. Furthermore, their reactive and often volatile nature poses difficulties on sample preparation in order to ensure the integrity of the analytes.
Therefore, the analyst should always decide on which is the correct analytical technique for each particular GTI determination. Among the five major alternatives (GC, LC, GC–MS, LC–MS and NMR), the GC-based techniques are preferred for the most volatile compounds, and the MS-based alternatives should be used for the cases requiring the highest sensitivity.
The chromatographic techniques are very complementary with each other. However, NMR has areas of application which can overlap with all of them, being most suitable when the compounds are not too volatile and their abundance is not too low [178].
The advantages, disadvantages and potentials for the use of NMR in GTI analysis have been recently discussed in depth [179]. Therefore, with the modern digitizers, the LOD and LOQ in qNMR are dependent on the number of scans, the solubility of the API and the strength of the magnetic field.
Although not many examples are currently available to illustrate the potential of NMR spectroscopy in this area, it is clear that this is one of the methods that will increase in importance in the coming years for detecting and quantitating these impurities.
A potential genotoxic impurity from the synthesis of an API was used to challenge the LOD/LOQ on a 400 MHz spectrometer with a limit test. Samples were spiked with the impurity up to 100 ppm and spectra were acquired (4096 scans, D1 = 2 s) with the inverse-gated 13C decoupling sequence to prevent overlapping satellites with the impurity. Unequivocal detection of the spiked genotoxic impurity was demonstrated at a level of 25 ppm [180]. In addition, qNMR was used for quantitation of methanosulfonic acid throughout the production process of an API and a practical quantification limit of 100 ppm was consistently attained [181].
Hydrazines, hydrazides and hydrazones must also be controlled in APIs [182]. Aryl hydrazone isomerization, resulting in the formation of residual geometrical E-isomer impurities, were determined by HPLC with DAD and MS–MS detection, followed by confirmatory NMR data [183].
Combined application of LC and NMR spectroscopy allowed characterization of mutagenic impurities in vestipitant. The starting material for the synthesis was identified as the root cause of impurities [184].
Finally, a toxic impurity was isolated from the experimental anti-neoplastic drug XP315. Characterization by combination of NMR (400 MHz, including gCOSY, gHMBC, gHSQC) and mass spectral methods revealed that it resulted from the dimerization of an intermediate by ring fusion, during the synthetic process [185].
2.1.7. Organic volatile impurities (residual solvents) and other accompanying impurities
The organic volatile impurities (OVIs) are a potential toxic risk for pharmaceutical products and have been a concern of manufacturers for many years. The ICH addresses this topic in its Q3C guideline [186]. The most widely used method for analysis of OVIs is GC [187]; however, there has been a slow but increasing use of NMR in this area. When the first coupled continuous-flow capillary GC–solenoidal microcoil 1H NMR spectroscopy (400 MHz) experiments were disclosed, the separation of a mixture of diethyl ether, dichloromethane and tetrahydrofuran, at an oven temperature of 60 °C was informed [69]. Acetone, resulting from the extraction process was identified and quantitated by NMR spectroscopy in dihydroquercetin, isolated from natural sources [90].
Preparation by-products such as ethanol and acetone are among the most common impurities identified in heparin preparations by 1H and 13C NMR spectroscopy [104], [188] and [189]. Being a process impurity, for the development of a chemometric-NMR approach to heparin analysis, the presence of EtOH was considered a potential interference for the determination of the drug or other impurities [106]. Other residual solvents like methanol, formic acid [104], and butanol (δ = 0.91, 1.35, 1.53, and 3.61 ppm) were also found in heparin samples [190], as well as sodium acetate and the chelating agent ethylenediaminetetraacetic acid (EDTA) (δ ∼ 3.5 ppm) [191].
It has been reported [192] that when initial studies concluded that direct injection or headspace GC could not be employed to determine the levels of N-methyl pyrrolidinone (NMP) in a process intermediate, a 1H NMR assay was developed and validated to directly quantitate the solvent at a 140 ppm level in the sample matrix.
Aldehydes and silicon derivatives have also been observed by NMR spectroscopy. Bortezomib is a modified dipeptidyl boronic acid [193]. Recent comparison between two brands of the drug revealed the presence of tert-butanol, a compounding solvent. In addition, this API proved to have silicon-containing impurities such as silanes and siloxanes and isovaleraldehyde as an impurity of unknown origin [194].
The related aldehydes, acetaldehyde and propionaldehyde, were quantitatively determined as impurities in poloxamer 188 by 1H NMR [195]. On the other hand, the NMR detection limit of residual silicone oil in poly(lactic acid) and poly(lactic acid–co-glycolic acid) microspheres, was determined to be 100 ppm, which compared favorably with IR determinations (5000 ppm) [196].
NMR spectroscopy was employed to determine the levels of residual benzene, toluene, acetone, and ethyl ether in cocaine [197]; ethyl acetate and methylene chloride, which were previously not detected in cocaine samples, were also found. An excellent correlation was obtained between the results of Schöniger flask combustion analysis for chlorine as an appraisal of chlorinated solvent levels and NMR-based estimates of chloroform. This demonstrated the suitability of NMR spectroscopy for the quantitation of chlorinated solvents [198].
2.2. Degradation products
2.2.1. Impurities resulting from degradation of synthetic APIs
Forced degradation is a useful tool for impurity profiling [199] and [200]. Due to its multiple advantages, NMR spectroscopy is widely used in pharmaceutical analysis in order to understand the course of the degradation and also identify and structurally elucidate its embedded impurities.
Stress tests of eprinomectin, a synthetic acetylated amino derivative of abamectin, revealed two impurities. Given their isomeric nature (2-epimer and Δ2,3-isomer) their structures became fully elucidated only after analysis of 1H, 13C, NOESY and COSY experiments [201].
The novel antiepileptic drug carisbamate undergoes significant degradation when subjected to stress conditions. The degradation products were isolated and characterized. MS/MS and 2D-NMR (COSY and HSQC) studies revealed that one of the degradation products resulted from positional isomerization of the carbamate moiety of the API [202], being prepared as shown in Scheme 4.
Full-size image (16 K)
Scheme 4.
Synthesis of carisbamate and origin of its isomeric impurity.
Figure options
Twelve impurities of clindamycin palmitate hydrochloride were isolated by preparative HPLC and characterized by a combination of LC–MS, FT-IR and simple 1D NMR (1H, 13C and DEPT) techniques[176]. Thorough NMR analysis was used for the characterization of an oxidation impurity of clopidogrel[203] and impurities of pridinol mesylate [204].
An impurity of citalopram was isolated by semi-preparative HPLC and its structure was established by MS, NMR and IR spectroscopy [142]. Analogously, chloroquine-N-oxide was identified as the major oxidative degradation product of chloroquine [205]. An N-oxidation product of cetirizine was isolated and characterized by 400 MHz NMR spectroscopy along with LC–MS/MS. The product is also a metabolite of the drug [206].
Two isomeric monoacylated diacerein impurities (MW = 326) were characterized by NMR spectroscopy, which confirmed their structures and revealed the exact position of the acetyl group in each case [207]. Unknown impurities of the antidepressant drug escitalopram were elucidated by 1H and 13C NMR and their structures were confirmed by synthesis [143]. Preparative HPLC, LC–MS/MS, UPLC–TOF-MS, NMR and FT-IR spectroscopy were employed to study the forced degradation of dipyridamole product, resulting in two new impurities (one of them an interaction product) which were characterized by NMR spectroscopy[208]. LC–NMR and LC–MS allowed to elucidate a new impurity in the antifungal drug icofungipen [209].
A novel impurity in eprosartan bulk drug was characterized by ESI/MSn and NMR [210], and five impurities were found in candesartan cilexetil subjected to stability and forced degradation studies. They were structurally elucidated employing NMR techniques [211]. Low-level unknown impurities in GW876008, a novel corticotrophin-release factor 1 antagonist were isolated and characterized employing LC–NMR and HR-NMR in conjunction with mass spectrometric experiments [212]. Similarly, degradation products of anastrozole [213] and etimicin sulfate were determined by LC–MS/MS and NMR spectroscopy and LC–ESI–MSn and NMR, respectively [158]. LC–NMR was employed for structural characterization of the oxidative degradation products of SCH 56592, an antifungal agent [214].
Two unknown impurities of moxidectin were isolated by flash chromatography during stress testing of the drug, and fully characterized by 1D (1H, 13C) and 2D (NOESY, COSY, HMBC and HSQC) NMR experiments [215].
The photodegradation of fleroxacin was investigated in different injections and solutions. The three major photodegradation products were detected and isolated by HPLC, and elucidated employing FT-IR, MSnand TOF-MS, as well as 1H, 13C, DEPT, and 2D NMR. Significant differences in the photodegradation process and photostability between the concentrated injection and the dilute injection have been demonstrated, and photo-degradation pathways were proposed [216].
Analogously, photo-irradiation of tablets containing pramipexole afforded pyrrolidine carboxylic acids (Impurities 1 and 2) and catechol (Impurity-6). The drug was also smoothly degraded in the methanolic solution yielding carboxylic esters; a possible degradation mechanism was proposed in which the reaction starts by a [2+2] cycloaddition of 1O2 to the thiazole ring (Scheme 5). Cleavage of the Csingle bondC bond of the thiazole, followed by attack of the secondary amine attack to the carbonyl group affords a pyrrolidinic structure, which is solvolyzed to furnish carboxylic acids and methyl esters depending on the reaction medium [217].
Full-size image (26 K)
Scheme 5.
Proposed photo-degradation mechanism of pramipexole.
Figure options
A photolytic degradation product of telmisartan and degradation products of irbesartan were detected and characterized using LC–MS/TOF, LC–MSn, LC–NMR and on-line H/D exchange mass studies[218] and [219].
The structure of the major degradation product of ezetimibe was initially proposed by NMR spectroscopy (1H and 13C) and MS studies [220]. Shortly after, and without providing a structure, it was pointed out that the previously proposed structure was inconsistent with the reported spectroscopic data [221]. Finally, a third group of scientists [222] effected detailed analysis of 1H, 13C, COSY, HSQC, HMBC and NOESY experiments, proposing a structure which agreed with a previously reported compound; 19F NMR was also employed in this research [223] and [224].
The thermal and alkaline degradation products of meropenem were identified by NMR and MS analysis. The in vitro cytotoxicity assay against mononuclear cells demonstrated their cytotoxicity, rising safety concerns and suggesting that special care must be taken to avoid exposure of the drug to the degradation conditions during the handling and storage of the pharmaceutical preparation [225].
Table 3 summarizes the degradation and process impurities found in some active pharmaceutical ingredients, while Table 4 contains a brief list of degradation products found by submission of different pharmaceutical ingredients to stress or forcing degradation conditions.
Table 3.Degradation and process impurities detected in some active pharmaceutical ingredients.
Drug Field (MHz) NMR Experiments Remarks Ref.
Asperosaponin VI 500 1H, 13C, HSQC, HMBC, COSY, ROESY, TOCSY Imp-1, Imp-2, Imp-5 and Imp-6: Natural impurities.
Imp-4: Resulting from acid hydrolysis. Formed by loss of a unit of arabinose.
Imp-7: Resulting from acid and basic hydrolysis. Formed by loss of two units of glucose.
Imp-3: Mixture of two epimers that are isomers of Asperosaponin VI. [172]
Bicalutamide 200 1H Imp-1 (I) and Imp-3 (III): Formed by basic degradation.
Imp-2 (II) and Imp-4 (IV): Side products.
Imp-5 (V): Starting material.
Imp-6 (VI) and Imp-7 (VIII): Process intermediates. [226]
Carisbamate 300 1H, 13C, COSY, HSQC Imp-1 – Imp-4: Process intermediates.
Imp-5 and Imp-6: Degradation products. [202]
Doxofylline 300 1H, 13C Imp-1: Starting material.
Imp-2–6: Process intermediates.
Imp-7 (DP-I): Resulting from acid degradation.
Imp-8 (DP-II): Produced by basic and acid degradation. [227]
Lopinavir 300 1H Imp-1 (RS1): Process intermediate.
Imp-2 (RS2): Starting material.
Imp-3 (RS3): Resulting from acid hydrolysis.
Imp-4 (RS4) and Imp-5 (RS5): Side products and thermal degradation products, formed on extended storage at higher temperature.
Imp-6 (RS6) and Imp-8(RS8): Impurities derived from the starting material.
Imp-9 (RS9) and Imp-10 (RS10): Side products. [100]
Table options
Table 4.Degradation products observed after exposing APIs or their products to stress conditions.
Drug Field (MHz) Experiments Remarks Ref.
Anastrozole 300 1H, 13C Imp-1: Formed by basic hydrolysis of both nitrile groups of the API (diacid).
Imp-2: Formed by basic hydrolysis of one nitrile group of the API (monoacid). [213]
Artesunate-Amodiaquine 500 1H, 13C, COSY, HSQC, HMBC, TOCSY, NOESY Imp-1: Rearranged derivative of anhydro dihydroartemisinin. [229]
Biapenem 500 1H, 13C, COSY, HSQC, HMBC Imp-1 – Imp-3: Products of hydrolysis.
Imp-2: Dimer A
Imp-3: Dimer B [230]
Chloroquine 500 1H, 13C, HSQC Imp-1 (Chloroquine N-oxide): Resulting from oxidative degradation. [205]
Clopidogrel 400 1H, 13C, DQF-COSY, HSQC Imp-1: Oxidation impurity.
Imp-2: Clopidogrel related compound A.
Imp-3 (Related compound B1) and Imp-4 (Related compound B2): Positional stereoisomers of the API.
Imp-5 (Related compound C): Optical isomer. [203]
Dipyridamole 400 1H, gDQCOSY, gHSQC, gHMBC Imp-1 (DPI): Formed by hydrolytic loss of a piperidine moiety resulting under acidic and oxidative (peroxide) conditions.
Imp-2 (DPII): Interaction product with tartaric acid formed during stress treatment under acidic, oxidative (peroxide) and thermal degradation conditions. [208]
Eprinomectin (EPM) 400 1H, 13C, NOESY, COSY Imp-1 (Unknown 1): Alkaline degradation product. Identified as the 2-epimer of EPM.
Imp-2 (Unknown 2): Alkaline degradation product of Imp-2. Identified as Δ20,03-EPM. [202]
Estradiol 500 1H, 13C Imp-1: Oxidative product of estradiol. [231]
Ezetimibe 400, 500 1H, 13C, gCOSY, gHSQC, gHMBC, 1D NOESY The impurity (2R,3R,6S)-N,6-bis(4-fluorophe nyl)-2-(4-hydroxy phenyl)-3,4,5,6-tetrahydro-2H-pyran-3-carboxamide was mis-identified in previous publications. [222]
Irbesartan 500 LC–NMR was used.
1H, COSY
(with solvent suppression) Imp-1 (DP-I): Produced by acid degradation.
Imp-2 (DP-II): Basic degradation product.
Imp-3 (DP-III): Resulting from photo-acid degradation. [219]
Meropenem 500 1H, 13C, COSY, HSQC Imp-1 (DP1): Thermal degradation product.
Imp-2 (DP2): Alkaline degradation product. [227]
Olmesartan medoxomil 600 1H, 13C, using a 1H–13C/15N triple resonance inverse probe in stopped-flow LC–NMR. Imp-1 (DP-1): Esterified dimer of Olmesartan. [232]
Pramipexole hydrochloride hydrate 400 1H, 13C, 2D (unspecified experiments) Imp-1: Photo-degradation product. [217]
Pridinol mesylate 300 1H, 13C Imp-1 (ELI): Dehydration product formed under acidic conditions.
Imp-2 (NOX): Formed under oxidative degradation conditions. [204]
Thiocolchicoside 300 1H, 13C Imp-1 (D1SO) and Imp-2 (D1SO2): Formed by oxidative degradation with m-CPBA.
Imp-3 – Imp-5 (D2, D3, D4): Acid degradation products. [233]
Table options
2.2.2. Impurities resulting from degradation of natural product APIs
Artesunate is the hemisuccinic ester of dihydroartemisinin, a product resulting from the reduction of artemisinin. When held under ICH zone IV storage conditions during 6 months (40 °C and 75% RH), artesunate is known to yield several products above the threshold of identification [228].
Artesunate-amodiaquine bilayer tablets were subjected to heat treatment (70 °C, 14–30 days, 75% RH), furnishing a new and degradation product (Impurity B). This was purified by flash chromatography followed by semipreparative HPLC and unequivocally identified by 500 MHz NMR experiments (especially COSY, TOCSY and HMBC) as the tetrahydrofuranyl acetate-rearranged derivative of anhydrodihydroartemisinin (Fig. 3); MS analysis completed the identification.
Full-size image (33 K)
Fig. 3.
Degradation of artesunate and three-bond H → C correlations observed in the HMBC spectrum of Impurity B.
Figure options
Further, an authentic standard of the impurity was synthesized. An analogous rearrangement has been described to occur for artemisinin congeners during a biotransformation process [234]. The product has been shown to be a surrogate secondary marker for the Fe(II)-catalyzed radical reaction to rearranged tetrahydrofuranyl acetate products [235] and [236].
An isomeric 2-deoxy-4α-hydroxy-anhydrodihydroartemisinin derivative was also isolated. This product, a result of intrinsic thermal unstability of artesunate itself, is produced by the homolytic radical opening of the endoperoxide bond, a transformation already recorded for other artemisinin derivatives [228].
Tacrolimus (FK506) is an immunosuppressive drug used to avoid organ rejection in patients that have undergone organ transplantation. This is a potent drug which has a narrow therapeutic index. Following clinical reports suggesting that use of generic brands resulted in a significant reduction in the tacrolimus concentration/dose ratio in the plasma of liver and kidney recipients, a group of analytical methods were employed to assess the level of impurities and potency of products found in the US marketplace [237]. Ascomycin has immunosuppressant properties similar to tacrolimus, which fundaments its determination. In addition, it has been reported that the drug tautomerizes, yielding related rearrangement products[238] and [239], and the analysis can be further complicated by the presence of mixtures of interconverting rotamers [240].
After preparing standards of tacrolimus, ascomycin and the excipients commonly found in the pharmaceutical formulations and performing the 1H NMR spectral comparisons, no ascomycin was detected above the LOD (0.7 w/w%) nor other known impurities were observed in any of the products. This clearly indicated that the clinically observed difference does not originate from quality (purity and potency) differences between the generic and innovator tacrolimus.
Even established APIs still hide secrets that await to be uncovered. The structure of a labile estradiol-related degradant, isolated from a pharmaceutical formulation containing estradiol and norethisterone acetate was elucidated by off-line and on-line NMR [241] as well as MS techniques. The product, which proved to be unstable, contains a 9,10-epoxy function. Its secondary and tertiary decomposition products were also identified, and a new oxidative degradation mechanism was proposed, which provides an improved explanation for the formation of some oxidative secosteroid degradation products [231]. The study also cleared some literature ambiguities.
Erythromycin is a complex macrolide antibiotic mainly composed of erythromycin A and also of erythromycins B-F. Several related substances may be formed by fermentation along with the aforementioned compounds. Unknown compounds found in an erythromycin formulation were investigated by LC coupled to MS and NMR. The degradation products were structurally elucidated, especially employing an array of off-line NMR techniques (1D 1H and 13C, and 2D TOCSY, NOESY, COSY, HSQC and HMBC); two of the degradation products were new [242].
2.3. Miscellaneous impurities
2.3.1. Impurities and degradation products related to excipients
Examples are scarce, and include degradation products of the excipients themselves and a case where an excipient promoted degradation of an API. Interaction products involving excipients are detailed separately. An unknown degradation product found in non-MS compatible HPLC analysis of a drug formulation was studied using a multidisciplinary approach. NMR analysis revealed it to be 5-hydroxymethyl furfural, resulting from degradation of lactose [243].
In addition, 1H NMR approaches to the determination of free fatty acids [244] and [245] and peroxidation products [246] in pharmaceutical lipids were recently reported. The first method was demonstrated in different fatty oils, fat samples, waxes and oleyloleat. The lipids are a group of widely used pharmaceutical excipients, many of which also have therapeutic properties [247] and [248].
A constituent of the artificial flavoring of a losartan potassium extemporaneous suspension formulation was held responsible for the photosensitized degradation of the API in the drug product [249]. The structures of the degradation products were determined using a combination of preparative HPLC, LC–MS and NMR (1H, 13C and 2D) experiments.
2.3.2. Leaching impurities
Low levels of extractables have been observed in vials of interferon-α2b finished product, following periods of filling line down time. Their structures were elucidated as chlorinated biphenyl derivatives by MS and NMR spectroscopic methods and their origin was found in the Rehau RAU-SIK silicone tubing used on the Bosch filling line [250]. The structures of these impurities revealed that they are formed during the polymerization process used to make the raw material for the tubes.
2.3.3. Interaction products
During a stability study of an enema formulation containing 5-aminosalycylic acid (5-ASA) and citric acid, the formation of three new peaks was observed by HPLC–MS and further investigated by HPLC–SPE–NMR. These proved to be citric acid amides of 5-ASA; therefore, a recommendation was issued for avoiding the use of citric acid in liquid 5-ASA formulations [251]. Impurities in 5-ASA have also been elucidated by LC–NMR [252].
Analogously, one of the impurities found after submission of a dipyridamole drug product preparation to stress conditions was a tartrate ester, resulting from esterification with tartaric acid, employed as an excipient [208].
Different pharmaceutical preparations against the common cold containing phenylephrine (PHE) and saccharose were studied and new tetrahydroisoquinoline-type impurities were discovered, arising from a phenolic cyclization between the API with sugar-derived formaldehyde (Scheme 6). These were characterized by LC–MS and NMR techniques [253].
Full-size image (15 K)
Scheme 6.
Formation of interaction products between phenylephrine and sugar-derived formaldehyde.
Figure options
An amide interaction product between phenylephrine and maleic acid in a combined cold formulation containing phenylephrine hydrochloride and chlorpheniramine maleate, has been reported NMR analysis confirmed the structure of the correct isomer formed, the α-hydroxyamide [254].
In addition, a hydroxylamine-type degradation compound of carvedilol drug product was characterized by NMR; its generation was attributed to an interaction of the drug with polyvinyl pyrrolidone (PVP) in the presence of moisture [255].
3. Other NMR active nuclei
Fluorine (19F) NMR spectra are invaluable in the analysis of fluorine containing compounds [256].When the impurity profile of 16 commercial formulations of ciprofloxacin was studied, and characterized using 19F and 1H NMR spectroscopy, it was found that four to twelve fluorinated impurities, among them fluoride ion and two already known compounds, were detected and quantified in the formulations analyzed by 19F NMR [257].
The 19F NMR analysis of five pharmaceutical preparations of fluoxetine and fluvoxamine revealed that one of the samples contained an unidentified fluorinated impurity [258]. NMR spectroscopy (1H, 13C, 19F,1H–1H, 1H–13C, HMBC) and especially a nOe experiment helped to elucidate the place where substitution of a fluorine by a dimethylamino group took place in fluconazole (Scheme 7), resulting in a new, monofluorinated impurity of the latter [259]. No changes were observed by NMR when the drug was subjected to ionizing radiation (20–200 kGy) [260]. The degradation product of a fluorinated thiazole-containing compound was identified by extensive application of NMR spectroscopy, including 19F and 15N techniques [261].
Full-size image (22 K)
Scheme 7.
Last step of the synthesis of fluconazole and generation of the monofluorinated impurity.
Figure options
19F Solid state NMR spectroscopy was associated with chemometrics in order to study the solid phases of atorvastatin. The proposed method allows recognizing the mixtures of the prepared forms of the API with a LOD of the crystalline phase impurities in the amorphous phase < 1% [262]. In addition, the distinction between monomeric and dimeric fluorinated impurities was carried out by 19F NMR spectroscopy [263]. The complete assignment of a mixture of isomers of 1,3-perfluorodimethylcyclohexane as well as some impurities were identified using a combination of 19F-COSY, 19F-TOCSY and 19F-NOESY experiments[264].
15N NMR spectroscopy has very low sensitivity; however, it has been employed for confirming the identity of impurities and drug degradation products [260]. A cationic impurity was identified in preladenant employing a series of 1D and 2D NMR experiments, including the gHMBCAD experiment for accessing1H–15N long-range correlation information [265] and the structure of an impurity in valsartan was confirmed with the aid of 15N NMR experiments [266].
4. Conclusions and perspectives
The presence of minor amounts of unwanted chemicals might influence the efficacy and safety of the pharmaceutical products. Therefore, their structures should be elucidated when present at level higher than 0.1%. These impurities are also synthesized based on the suggested structures, as standards for selective analytical methods for their quantitation in drug substance and products.
NMR spectroscopy is finding increasing application for the identification of unknown organic impurities embedded in pharmaceutical preparations. In addition, this is the most suitable technique for the identification and the qualitative analysis of mixtures, where some of the compounds are unknown, and almost unsubstitutable for structural analysis of epimers, rotamers and even some cases of enantiomers. NMR is highly informative and it allows to draw conclusions on the structure of the components in a mixture of related compounds because the NMR spectrum is sensitive to even small modifications in their molecular geometry or bonding.
NMR spectroscopy is usually employed to obtain structural information. In some cases, it is used to confirm structural proposals made as a result of MS experiments, since the combination of NMR and MS is one of the most powerful approaches to acquire complete information on the structure of a studied molecule or mixture of compounds.
The quantitative determination of the level of impurities is one of the major applications of qNMR in this area. This can be done along with acquisition of their structural information. The determination of related impurities in a mixture, as well as content of residual solvent, isomeric composition, molar ratio and even the course of a degradation can be also achieved.
It has been shown that LC–NMR ranks among the most powerful hyphenated techniques for the separation and structure elucidation of complex organic molecules, also providing quantitative information. Due to the success of LC–NMR hyphenation, it can be foreseen that this approach will be extended to other multidimensional separation schemes in the near future. Despite the number of years of their evolution, coupled NMR techniques are still in their infancy and will require much more time to become established. Particularly, GC–NMR could be highly useful for volatiles. It is also expected that instrumental improvements will make more feasible the coupling of fractionation with 13C NMR detection.
The relatively low sensitivity, sometimes poor reproducibility and high instrumental costs are current substantial limitations of this spectroscopy. State-of-the-art NMR equipment, including higher magnetic fields and cryo-probes exhibit enhanced sensitivity. However, despite the latest advances, it should be stressed that current sensitivity still makes NMR unsuitable for quantitative analysis of most of the genotoxic impurities, even employing the most sensitive 1H NMR techniques.
Conquering all these technological frontiers required for trace and impurity analysis will open a completely new area of exploration and research in pharmaceutical analysis.
Acknowledgements
The authors gratefully acknowledge Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), Secretaría de Ciencia y Técnica (SECyT-UNR) and Secretaría de Ciencia, Tecnología e Innovación (SECTeI) for financial support. NLC Thanks CONICET for her fellowship.
Appendix A. Acronyms, abbreviations and their definitions, regarding NMR experiments
Acronyma/abbreviation Meaning
1D, 1D-NMR Mono-dimensional NMR experiment
1H qNMR, qHNMR Quantitative proton NMR experiment
2D, 2D-NMR Two-dimensional NMR experiment
APT Attached Proton Test
COSY Homonuclear Correlation Spectroscopy
DBPPSTE DOSY DOSY Bipolar Pulse Pair Stimulated Echo
DEPT Distortionless Enhancement by Polarization Transfer
DOSY Diffusion Ordered Spectroscopy
DQF-COSY Double Quantum Filtered COSY
HETCOR Heteronuclear Shift Correlation
HMBC Heteronuclear Multiple-Bond Correlation
HMQC Heteronuclear Multiple-Quantum Correlation
HSQC Heteronuclear Single Quantum Correlation
NOE Nuclear Overhauser Effect
NOESY Nuclear Overhauser Effect Spectroscopy
qNMR Quantitative NMR Spectroscopy
ROESY Rotating-Frame NOE Spectroscopy
TOCSY Total Correlation Spectroscopy
zTOCSY z-Filtered Total Correlation Spectroscopy
a
Prefix and suffix letters are added to more elaborate experiments. Letter ‘g’ is employed to denote the field gradient version of the experiment. The suffix ‘AD’ indicates the use of adiabatic pulses.
Table options
References
[1]
M. Sautel, A.M. Krstulovic
Pharmaceutical analysis beyond compendial requirements
J. Sep. Sci., 28 (28) (2005), pp. 1538–1629
[2]
S. Ahuja, K.M. Alsante (Eds.), Handbook of Isolation and Characterization of Impurities in Pharmaceuticals, Elsevier, Amsterdam, The Netherlands (2003)
[3]
S. Husain, R. Nageswara Rao
Monitoring of process impurities in drugs
Z. Deyl, I. Miksik, F. Tagliero, E. Tesarová (Eds.), Advanced Chromatographic and Electromigration Methods in Biosciences, Elsevier, Amsterdam, The Netherlands (1998), pp. 833–888
Article
|
PDF (2179 K)
|
View Record in Scopus
| | |
Citing articles (1)
[4]
K.M. Alsante, R.C. Friedmann, T.D. Hatajik, L.L. Lohr, T.R. Sharp, K.D. Snyder, E.J. Szczesny
Degradation and impurity analysis for pharmaceutical drug candidates
S. Ahuja, S. Scypinski (Eds.), Handbook of Modern Pharmaceutical Analysis, Academic Press, San Diego, USA (2001), pp. 85–172
Article
|
PDF (5199 K)
|
View Record in Scopus
| | |
Citing articles (13)
[5]
C. Todd, E. Sheinin
S. Ahuja, M. Dong (Eds.), Handbook of Pharmaceutical Analysis by HPLC, Academic Press, San Diego, USA (2005), pp. 359–377
View Record in Scopus
| | |
Citing articles (1)
[6]
L.Z. Zhou
S. Ahuja, M. Dong (Eds.), Handbook of Pharmaceutical Analysis by HPLC, Academic Press, San Diego, USA (2005), pp. 499–568
Article
|
PDF (7837 K)
|
View Record in Scopus
| | |
Citing articles (3)
[7]
J.C. Berridge
Impurities in drug substances and drug products: new approaches to quantification and qualification
J. Pharm. Biomed. Anal., 14 (1995), pp. 7–12
Article
|
PDF (479 K)
|
View Record in Scopus
| | |
Citing articles (25)
[8]
A.M. Krstulovic, C.R. Lee
Defining drug purity through chromatographic and related methods: current status and perspectives
J. Chromatogr. B, 689 (1997), pp. 137–153
Article
|
PDF (859 K)
|
View Record in Scopus
| | |
Citing articles (20)
[9]
R.N. Rao, V. Nagaraju
An overview of the recent trends in development of HPLC methods for determination of impurities in drugs
J. Pharm. Biomed. Anal., 33 (2003), pp. 335–377
[10]
J.V. Rompay
Purity determination and evaluation of new drug substances
J. Pharm. Biomed. Anal., 4 (1986), pp. 725–732
[11]
S. Ahuja
Assuring quality of drugs by monitoring impurities
Adv. Drug Deliv. Rev., 59 (2007), pp. 3–11
Article
|
PDF (227 K)
|
View Record in Scopus
| | |
Citing articles (35)
[12]
S. Ahuja
Impurities Evaluation of Pharmaceutical
Marcel Dekker, New York, USA (1998)
[13]
S. Görög
Identification and Determination of Impurities in Drugs
Elsevier, Amsterdam, The Netherlands (2000)
[14]
M. Webb, R. Smith (Eds.), Analysis of Drug Impurities, Blackwell, Oxford UK (2007)
[15]
N. Rahman, S.N. Hejaz Azmi, H.-F. Wu
The importance of impurity analysis in pharmaceutical products: an integrated approach
Accred. Qual. Assur., 11 (2006), pp. 69–74
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (11)
[16]
D. Bartos, S. Görög
Recent advances in the impurity profiling of drugs
Curr. Pharm. Anal., 4 (2008), pp. 215–230
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (33)
[17]
S.B.K.B. Bari, Y.S. Jaiswal, A.A. Shikhedkar
Impurity profile: significance in active pharmaceutical ingredient
Eurasian J. Anal. Chem., 2 (2007), pp. 32–53
View Record in Scopus
| | |
Citing articles (32)
[18]
B. De Spiegeleer, V. Vergote, A. Pezeshki, K. Peremans, C. Burvenich
Impurity profiling quality control testing of synthetic peptides using liquid-chromatography-photodiode array fluorescence and liquid chromatography–electrospray ionization-mass spectrometry. The obestatin case
Anal. Biochem., 376 (2008), pp. 229–234
Article
|
PDF (277 K)
|
View Record in Scopus
| | |
Citing articles (36)
[19]
S.W. Baertschi
Analytical methodologies for discovering and profiling degradation-related impurities
Trends Anal. Chem., 25 (2006), pp. 758–767
Article
|
PDF (161 K)
|
View Record in Scopus
| | |
Citing articles (28)
[20]
I.C.H.
Impurities in new drug substances Q3A (R2)
International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2006)
[21]
ICH
Impurities in new drug products Q3B(R2)
International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2006)
[22]
C. Foti, K. Alsante, G. Cheng, T. Zelesky, M. Zell
Tools and workflow for structure elucidation of drug degradation products
Trends Anal. Chem., 49 (2013), pp. 89–99
Article
|
PDF (1269 K)
|
View Record in Scopus
| |
Citing articles (3)
[23]
K. Wilczewska, A. Kot-Wasik, J. Namieśnik
LC–MS and LC–NMR as complementary techniques for the determination of pharmaceuticals in dosage formulations
Crit. Rev. Anal. Chem., 43 (2013), pp. 148–175
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (2)
[24]
S. Singh, T. Handa, M. Narayanam, A. Sahu, M. Junwal, R.P. Shah
A critical review on the use of modern sophisticated hyphenated tools in the characterization of impurities and degradation products
J. Pharm. Biomed. Anal., 69 (2012), pp. 148–173
Article
|
PDF (1599 K)
|
View Record in Scopus
| |
Citing articles (38)
[25]
J. Chin, J.B. Fell, M. Jarosinski, M.J. Shapiro, J.R. Wareing
HPLC/NMR in combinatorial chemistry
J. Org. Chem., 63 (1998), pp. 386–390
Full Text via CrossRef
[26]
H. Thiele, G. McLeod, M. Niemitz, T. Kühn
Structure verification of small molecules using mass spectrometry and NMR spectroscopy
Monatsh. Chem., 142 (2011), pp. 717–730
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5)
[27]
S.K. Bharti, R. Roy
Quantitative 1H NMR spectroscopy
Trends Anal. Chem., 35 (2012), pp. 5–26
Article
|
PDF (788 K)
|
View Record in Scopus
| |
Citing articles (40)
[28]
M. Findeisen, S. Berger
50 and More Essential NMR Experiments: A Detailed Guide
Wiley, New York, USA (2013)
[29]
S. Berger, S. Braun
200 and More NMR Experiments
Wiley, New York, USA (2004)
[30]
G.A. Webb (Ed.), Modern Magnetic Resonance, Springer, Heidelberg, Germany (2007)
[31]
J. Keeler
Understanding NMR Spectroscopy
Wiley, New York, USA (2013)
[32]
H. Heise, S. Matthews (Eds.), Modern NMR Methodology, Springer, Heidelberg, Germany (2013)
[33]
N.E. Jacobsen
NMR Spectroscopy Explained: Simplified Theory, Applications and Examples for Organic Chemistry and Structural Biology
Wiley, New York, USA (2007)
[34]
M.Y. Iqbal, K.M.V. Narayana Rao, G. Sridhar, P. Padmanabha Raju, G.R. Deshpande, J.M. Babu
Characterization and relative response factor determination of process related impurity in naproxen by nuclear magnetic resonance spectroscopy
J. Pharm. Biomed. Anal., 56 (2011), pp. 484–490
Article
|
PDF (846 K)
|
View Record in Scopus
| |
Citing articles (4)
[35]
U. Holzgrabe, R. Deubner, C. Schollmayer, B. Waibel
Quantitative NMR spectroscopy – applications in drug analysis
J. Pharm. Biomed. Anal., 38 (2005), pp. 806–812
Article
|
PDF (262 K)
|
View Record in Scopus
| |
Citing articles (74)
[36]
M. Malet-Martino, U. Holzgrabe
NMR techniques in biomedical and pharmaceutical analysis
J. Pharm. Biomed. Anal., 55 (2011), pp. 1–15
Article
|
PDF (753 K)
|
View Record in Scopus
| |
Citing articles (40)
[37]
D.W.T. Claridge
High Resolution NMR Techniques in Organic Chemistry
Elsevier, Amsterdam, The Netherlands (2009)
[38]
S. Ito, T. Miki, M. Yoshikawa, M. Hamada, Y. Kawate, S. Hayashi, A. Sato, T. Kiyoshi, F. Matsumoto, H. Nagai, H. Wada, S. Fukui, T. Noguchi
Test restults of long term operation of the superfluid cooled cryostat for a 1 GHz NMR Spectrometer
IEEE Trans. Appl. Supercond., 12 (2002), pp. 1347–1350
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4)
[39]
P.F. Flynn, D.L. Mattiello, H.D.W. Hill, A.J. Wand
Optimal use of cryogenic probe technology in NMR studies of proteins
J. Am. Chem. Soc., 122 (2000), pp. 4823–4824
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (65)
[40]
H. Nagai, A. Sato, T. Kiyoshi, F. Matsumoto, H. Wada, S. Ito, T. Miki, M. Yoshikawa, Y. Kawate, S. Fukui
Development and testing of superfluid-cooled 900 MHz NMR magnet
Cryogenics, 41 (2001), pp. 623–630
Article
|
PDF (779 K)
|
View Record in Scopus
| |
Citing articles (8)
[41]
Y.Q. Song, B.M. Goodson, A. Pines
NMR and MRI using laser-polarized xenon
Spectroscopy, 14 (1999), pp. 26–33
[42]
U. Holzgrabe, B. Diehl, I. Wawer (Eds.), NMR Spectroscopy in Pharmaceutical Analysis, Elsevier, Amsterdam, The Netherlands (2008)
[43]
A. Karioti, E. Giocaliere, C. Guccione, G. Pieraccini, E. Gallo, A. Vannaccic, A.R. Bilia
Combined HPLC–DAD–MS, HPLC–MSn and NMR spectroscopy for quality control of plant extracts: the case of a commercial blend sold as dietary supplement
J. Pharm. Biomed. Anal., 88 (2014), pp. 7–15
Article
|
PDF (2222 K)
|
View Record in Scopus
| |
Citing articles (3)
[44]
I. McEwen, A. Elmsjö, A. Lehnström, A. Hakkarainen, B.M. Johansson
Screening of counterfeit corticosteroid in creams and ointments by NMR spectroscopy
J. Pharm. Biomed. Anal., 70 (2012), pp. 245–250
Article
|
PDF (575 K)
|
View Record in Scopus
| |
Citing articles (6)
[45]
T. Beyer, B. Diehl, U. Holzgrabe
Quantitative NMR spectroscopy of biologically active substances and excipients
Bioanal. Rev., 2 (2010), pp. 1–22
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (22)
[46]
S. Görög, C. Szántay Jr.
Spectroscopic methods in drug quality control and development
J. Lindon, G. Tranter, D. Koppenaal (Eds.), Encyclopedia of Spectroscopy and Spectrometry (2nd ed.), vol. 3Elsevier, Oxford, UK (2010), pp. 2640–2650
Article
|
PDF (537 K)
|
View Record in Scopus
| |
Citing articles (5)
[47]
C. Szántay Jr., Z. Béni, G. Balogh, T. Gáti
The changing role of NMR spectroscopy in off-line impurity identification: a conceptual view
Trends Anal. Chem., 25 (2006), pp. 806–820
Article
|
PDF (316 K)
|
View Record in Scopus
| |
Citing articles (21)
[48]
C. Szántay Jr., A. Demeter
NMR spectroscopy
S. Görög (Ed.), Identification and Determination of Impurities in Drugs (1st ed.), Elsevier, Amsterdam, The Netherlands (2000), pp. 109–143
[49]
A. Aranyi
Isolation of impurities by (semi)preparative HPLC
S. Görög (Ed.), Identification and Determination of Impurities in Drugs (1st ed.), Elsevier, Amsterdam, The Netherlands (2000), pp. 240–251
Article
|
PDF (537 K)
|
View Record in Scopus
| |
Citing articles (3)
[50]
G.F. Pauli, T. Gödecke, B.U. Jaki, D.C. Lankin
Quantitative 1H NMR. Development and potential of an analytical method: an update
J. Nat. Prod., 75 (2012), pp. 834–851
View Record in Scopus
| |
Citing articles (70)
[51]
T. Ründlof, T.R. McEwen, M. Johansson, T. Arvidsson
Use and qualification of primary and secondary standards employed in quantitative 1H NMR spectroscopy of pharmaceuticals
J. Pharm. Biomed. Anal. (2013), p. 010 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.2013.09
[52]
M. Weber, C. Hellriegel, A. Rück, J. Wuethrich, P. Jenks
Using high-performance 1H NMR (HP-qNMR®) for the certification of organic reference materials under accreditation guidelines – describing the overall process with focus on homogeneity and stability assessment
J. Pharm. Biomed. Anal. (2013), p. 007 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.2013.09
View Record in Scopus
| |
Citing articles (1)
[53]
H. Jancke, F. Malz, W. Haesselbarth
Structure analytical methods for quantitative reference applications
Accred. Qual. Assur., 10 (2005), pp. 421–429
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (22)
[54]
T. Schoenberger
Determination of standard sample purity using the high-precision 1H-NMR process
Anal. Bioanal. Chem., 403 (2012), pp. 247–254
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (14)
[55]
F. Malz, H. Jancke
Validation of quantitative NMR
J. Pharm. Biomed. Anal., 38 (2005), pp. 813–823
Article
|
PDF (335 K)
|
View Record in Scopus
| |
Citing articles (206)
[56]
U. Holzgrabe
Quantitative NMR spectroscopy in pharmaceutical applications
Prog. Nucl. Magn. Reson. Spectrosc., 57 (2010), pp. 229–240
Article
|
PDF (1009 K)
|
View Record in Scopus
| |
Citing articles (74)
[57]
S. Mahajan, I.P. Singh
Determining and reporting purity of organic molecules: why qNMR
Magn. Reson. Chem., 51 (2013), pp. 76–81
View Record in Scopus
| |
Citing articles (10)
[58]
R. Nogueira, B.C. Garrido, R.M. Borges, G.E.B. Silva, S.M. Queiroz, V.S. Cunha
Development of a new sodium diclofenac certified reference material using the mass balance approach and 1H qNMR to determine the certified property value
Eur. J. Pharm. Sci., 48 (2013), pp. 502–513
Article
|
PDF (940 K)
|
View Record in Scopus
| |
Citing articles (7)
[59]
M. Weber, C. Hellriegel, A. Rück, R. Sauermoser, J. Wüthrich
Using high-performance quantitative NMR (HP-qNMRW) for certifying traceable and highly accurate purity values of organic reference materials with uncertainties < 0.1%
Accred. Qual. Assur., 18 (2013), pp. 91–98
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12)
[60]
U. Holzgrabe
Quantitative NMR spectroscopy in pharmaceutical applications
Progr. NMR Spectrosc., 57 (2010), pp. 229–240
Article
|
PDF (1009 K)
|
View Record in Scopus
[61]
R. Deubner, U. Holzgrabe
Micellar electrokinetic capillary chromatography, high performance liquid chromatography and nuclear magnetic resonance-three orthogonal methods for characterization of critical drugs
J. Pharm. Biomed. Anal., 35 (2004), pp. 459–467
Article
|
PDF (387 K)
|
View Record in Scopus
| | |
Citing articles (25)
[62]
K. Albert (Ed.), On-Line LC–NMR and Related Techniques, Wiley, New York USA (2002)
[63]
M.V. Silva Elipe
LC–NMR and Other Hyphenated NMR Techniques: Overview and Applications
Wiley, Hoboken, USA (2012)
[64]
Y. Kazakevich, R. LoBrutto (Eds.), HPLC for Pharmaceutical Scientists, Wiley, New York, USA (2007), pp. 901–936
[65]
N.C. Gonella
LC–NMR: Expanding the Limits of Structure Elucidation
CRC, Boca Raton, USA (2013)
[66]
M.-H. Wann
20 Application of LC–NMR in pharmaceutical analysis
Sep. Sci. Technol., 6 (2005), pp. 569–579
Article
|
PDF (532 K)
|
View Record in Scopus
| | |
Citing articles (7)
[67]
J.R. Kesting, K.T. Johansen, J.W. Jaroszewski
Hyphenated NMR techniques
A.J. Dingley, S.M. Pascal (Eds.), Advances in Biomedical Spectroscopy, Volume 3: Biomolecular NMR Spectroscopy, IOP Press, Amsterdam, The Netherlands (2011), pp. 413–434
View Record in Scopus
| | |
Citing articles (4)
[68]
M. Kühnle, D. Kreidler, H. Czesla, K. Holtin, P. Schuler, W. Schaal, V. Schurig, K. Albert
On-line coupling of gas chromatography to nuclear magnetic resonance spectroscopy: a method for the analysis of volatile stereoisomers
Anal. Chem., 80 (2008), pp. 5481–5486
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (18)
[69]
M. Grynbaum, D. Kreidler, J. Rehbein, A. Purea, P. Schuler, W. Schaal, H. Czesla, A. Webb, V. Schurig, K. Albert
Hyphenation of gas chromatography to microcoil 1H nuclear magnetic resonance spectroscopy
Anal. Chem., 79 (2007), pp. 2708–2713
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (25)
[70]
K. Pusecker, J. Schewitz, P. Gfrorer, L.-H. Tseng, K. Albert, E. Bayer, I.D. Wilson, N.J. Bailey, G.B. Scarfe, J.K. Nicholson, L.C. Lindon
On-flow identification of metabolites of paracetamol from human urine using directly coupled CZE-NMR and CEC-NMR spectroscopy
Anal. Commun., 35 (1998), pp. 213–215
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (41)
[71]
J. Schewitz, P. Gfrorer, K. Pusecker, L.-H. Tseng, K. Albert, E. Bayer, I.D. Wilson, N.J. Bailey, G.B. Scarfe, J.K. Nicholson, J.C. Lindon
Directly coupled CZE-NMR and CEC-NMR spectroscopy of metabolite analysis: paracetamol metabolites in human tissue
Analyst, 12 (1998), pp. 2835–2837
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (44)
[72]
K. Pusecker, J. Schewitz, P. Gfrorer, L.-H. Tseng, K. Albert, E. Bayer
On-line coupling of capillary electrochromatography, capillary electrophoresis, and capillary HPLC with nuclear magnetic resonance spectroscopy
Anal. Chem., 70 (1998), pp. 3280–3285
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (93)
[73]
P. Gfrorer, L.-H. Tseng, E. Rapp, K. Albert, E. Bayer
Influence of pressure upon coupling pressurized capillary electrochromatography with nuclear magnetic resonance spectroscopy
Anal. Chem., 73 (2001), pp. 3234–3239
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (49)
[74]
P. Gfrorer, J. Schewitz, K. Pusecker, L.-H. Tseng, K. Albert, E. Bayer
Gradient elution capillary electrochromatogrpahy and hyphenation with nuclear magnetic resonance
Electrophoresis, 20 (1999), pp. 3–8
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (51)
[75]
M.J. Little, N. Aubry, M.-E. Beaudoin, N. Goudreau, S.R. LaPlante
Quantifying trifluoroacetic acid as a counterion in drug discovery by 19F NMR and capillary electrophoresis
J. Pharm. Biomed. Anal., 43 (2007), pp. 1324–1330
Article
|
PDF (187 K)
|
View Record in Scopus
| | |
Citing articles (14)
[76]
D.A. Jayawickrama, J.V. Sweedler
Hyphenation of capillary separations with nuclear magnetic resonance spectroscopy
J. Chromatogr. A, 1000 (2003), pp. 819–840
Article
|
PDF (1199 K)
|
View Record in Scopus
| | |
Citing articles (50)
[77]
N. Wu, T.L. Peck, A.G. Webb, R.L. Magin, J.V. Sweedler
Nanoliter volume sample cells for 1H NMR application to on-line detection in capillary electrophoresis
J. Am. Chem. Soc., 116 (1994), pp. 7929–7930
Full Text via CrossRef
[78]
J. Schewitz, R. Pusecker, P. Gfrorer, U. Gotz, L.-H. Tseng, K. Albert, E. Bayer
Direct coupling of capillary electrophoresis and nuclear magnetic resonance spectroscopy for the identification of a dinucleotide
Chromatographia, 50 (1999), pp. 333–337
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (27)
[79]
M. Kühnle, D. Kreidler, K. Holtin, H. Czesla, P. Schuler, V. Schurig, K. Albert
Online coupling of enantioselective capillary gas chromatography with proton nuclear magnetic resonance spectroscopy
Chirality, 22 (2010), pp. 808–812
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (8)
[80]
P. Novak, M. Cindric, P. Tepes, S. Dragojevic, M.K. Ilija
Mihaljevic, Identification of impurities in acarbose by using an integrated liquid chromatography-nuclear magnetic resonance and liquid chromatography-mass spectrometry approach
J. Sep. Sci., 28 (2005), pp. 1442–1447
View Record in Scopus
|
Full Text via CrossRef
| | |
Citing articles (11)
[81]
F. Xu, A.J. Alexander
The design of an on-line semi-preparative LC-SPE-NMR system for trace analysis
Magn. Reson. Chem., 43 (2005), pp. 776–782
View Record in Scopus
| | |
Citing articles (31)
[82]
J.R. Kesting, J. Huang, D. Sorensen
Identification of adulterants in a Chinese herbal medicine by LC-HRMS and LC-MS-SPE/NMR and comparative in vivo study with standards in a hypertensive rat model
J. Pharm. Biomed. Anal., 51 (2010), pp. 705–711
Article
|
PDF (344 K)
|
View Record in Scopus
| |
Citing articles (27)
[83]
T. Murakami, N. Fukutsu, J. Kondo, T. Kawasaki, F. Kusu
Application of liquid chromatography-two-dimensional nuclear magnetic resonance spectroscopy using pre-concentration column trapping and liquid chromatography–mass spectrometry for the identification of degradation products in stressed commercial amlodipine maleate tablets
J. Chromatogr. A, 1181 (2008), pp. 67–76
Article
|
PDF (1021 K)
|
View Record in Scopus
| |
Citing articles (22)
[84]
J.K. Roberts, R.J. Smith
Use of liquid chromatography-nuclear magnetic resonance spectroscopy for the identification of impurities in drug substances
J. Chromatogr. A, 677 (1994), pp. 385–389
Article
|
PDF (380 K)
|
View Record in Scopus
| |
Citing articles (44)
[85]
I.N. Glazkov, N.L. Bochkareva, I.A. Revel’skii
Determination of organic impurities in pharmaceutical preparations
J. Anal. Chem., 60 (2005), pp. 107–118
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12)
[86]
M. Babjak, G. Balogh, M. Gazdag, S. Gorog
Estimation of impurity profiles of drugs and related materials. Part XXI. HPLC/UV/MS study of the impurity profile of ethynodiol diacetate
J. Pharm. Biomed. Anal., 29 (2002), pp. 1153–1157
Article
|
PDF (112 K)
|
View Record in Scopus
| |
Citing articles (14)
[87]
F. Guo, H. Feng, Y. Wang, C. Zhang, Y. Li
Characterization of related impurities in megestrol acetate
J. Pharm. Biomed. Anal., 41 (2006), pp. 1418–1422
Article
|
PDF (252 K)
|
View Record in Scopus
| |
Citing articles (4)
[88]
J. Hammond, I. Jones
The use of chromatography and online structure elucidation using spectroscopy
R.J. Smith, M.L. Webb (Eds.), Analysis of Drug Impurities, Blackwell Publishing, Oxford, UK (2007)
[89]
N.P. Mishchenko, S.A. Fedoreev, V.P. Glazunov, V.A. Denisenko, N.P. Krasovskaya, L.I. Glebko, L.G. Maslov, P.S. Dmitrenok, V.L. Bagirova
Isolation and identification of impurities in the parent substance of echinochrome and in the drug histochrome
Pharm. Chem. J., 38 (2004), pp. 50–53
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4)
[90]
V.V. Podgorskii, A.S. Mikhalev, G.A. Kalabin
Quantitative NMR spectroscopy for quality control of drugs and pharmaceuticals
Pharm. Chem. J., 45 (2011), pp. 194–197
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5)
[91]
Y. Peng, J. Luo, Q. Lu, X. Chen, Y. Xie, L. Chen, W. Yang, S. Du
HPLC analysis, semi-preparative HPLC preparation and identification of three impurities in salidroside bulk drug
J. Pharm. Biomed. Anal., 49 (2009), pp. 828–832
Article
|
PDF (357 K)
|
View Record in Scopus
| |
Citing articles (15)
[92]
B. Wang, H. Wang, E. Wang, W. Yan, W. Ye, P. Li
Isolation and structure characterization of related impurities in sodium tanshinone IIA sulfonate by LC/ESI-MSn and NMR
J. Pharm. Biomed. Anal., 67–68 (2012), pp. 36–41
Article
|
PDF (488 K)
|
View Record in Scopus
| |
Citing articles (3)
[93]
Q.G. Zou, B.Y. Huang, T.T. He, P. Wei, Z.J. Zhang, P.K. Ouyang
Identification, isolation, and characterization of impurities in sodium tanshinone IIA sulfonate, J
Liq. Chromatogr. Relat. Technol., 32 (2009), pp. 2346–2360
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (4)
[94]
Z. Dubrovay, V. Háda, Z. Bénia, C. Szántay Jr.
NMR and mass spectrometric characterization of vinblastine, vincristine and some new related impurities – Part I
J. Pharm. Biomed. Anal., 84 (2013), pp. 293–308
Article
|
PDF (1379 K)
|
View Record in Scopus
| |
Citing articles (4)
[95]
V. Háda, Z. Dubrovay, A. Lakó-Futó, J. Galambos, Z. Gulyás, A. Aranyi, C. Szántay Jr.
NMR and mass spectrometric characterization of vinblastine, vincristine and some new related impurities – Part II
J. Pharm. Biomed. Anal., 84 (2013), pp. 309–322
Article
|
PDF (1384 K)
|
View Record in Scopus
[96]
X. Cao, Y. Tai, C. Sun, K. Wang, Y. Pan
Characterization of impurities in semi-synthetic vinorelbine bitartrate by HPLC-MS with mass spectrometric shift technique
J. Pharm. Biomed. Anal., 39 (2005), pp. 39–45
Article
|
PDF (189 K)
|
View Record in Scopus
| |
Citing articles (4)
[97]
K. Petritis, C. Elfakir, M. Dreux
A comparative study of commercial liquid chromatographic detectors for the analysis of underivatized amino acids
J. Chromatogr. A, 961 (2002), pp. 9–21
Article
|
PDF (454 K)
|
View Record in Scopus
| |
Citing articles (108)
[98]
U. Holzgrabe, C.J. Nap, T. Beyer, S. Almeling
Alternatives to amino-acid-analysis for the purity control of pharmaceutical grade L-alanine
J. Sep. Sci., 33 (2010), pp. 2402–2410
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (12)
[99]
S. Kopec, U. Holzgrabe
Impurity profile of amino acids?
Pharmeurope Sci. Notes, 17 (2005), pp. 39–45
View Record in Scopus
| |
Citing articles (6)
[100]
S. Rao Chitturi, C. Bharathia, A.V. Raghava Reddy, K. Chandrasekhar Reddy, H. Kumar Sharma, V. Kumar Handa, R. Dandala, V.H. Bindu
Impurity profile study of lopinavir and validation of HPLC method for the determination of related substances in lopinavir drug substance
J. Pharm. Biomed. Anal., 48 (2008), pp. 1430–1440
[101]
H. Liu, Z. Zhang, R.J. Linhardt
Lessons learned from the contamination of heparin
Nat. Prod. Rep., 26 (2009), pp. 313–321
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (132)
[102]
S. Beni, J.F.K. Limtiaco, C.K. Larive
Analysis and characterization of heparin impurities
Anal. Bioanal. Chem., 399 (2011), pp. 527–539
[SD-008]
[103]
USP
Heparin Sodium, United States Pharmacopeia-National Formulary, USP 35-NF 30
Rand McNally, Rockville, USA (2013), pp. 3040–3042 3403–3406
[SD-008]
[104]
T. Beyer, M. Matz, D. Brinz, O. Rädler, B. Wolf, J. Norwig, K. Baumann, S. Alban, U. Holzgrabe
Composition of OSCS-contaminated heparin occuring in 2008 in batches on the german market
Eur. J. Pharm. Sci., 40 (2010), pp. 297–304
[SD-008]
[105]
H. Ye, T.K. Toby, C.D. Sommers, H. Ghasriani, M.L. Trehy, W. Ye, R.E. Kolinski, L.F. Buhse, A. Al-Hakim, D.A. Keire
Characterization of currently marketed heparin products: key tests for LMWH quality assurance
J. Pharm. Biomed. Anal., 85 (2013), pp. 99–107
[SD-008]
[106]
Q. Zang, D.A. Keire, R.D. Wood, L.F. Buhse, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
Combining, (1)H NMR spectroscopy and chemometrics to identify heparin samples that may possess dermatan sulfate (DS) impurities or oversulfated chondroitin sulfate (OSCS) contaminants
J. Pharm. Biomed. Anal., 54 (2011), pp. 1020–1029
[SD-008]
[107]
P.A.J. Mourier, O.Y. Guichard, F. Herman, C. Viskov
Heparin sodium compliance to USP monograph: structural elucidation of an atypical 2.18 ppm NMR signal
J. Pharm. Biomed. Anal., 67–68 (2012), pp. 169–174
[SD-008]
[108]
Q. Zang, D.A. Keire, L.F. Buhse, R.D. Wood, D.P. Mital, S. Haque, S. Srinivasan, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
Identification of heparin samples that contain impurities or contaminants by chemometric pattern recognition analysis of proton NMR spectral data
Anal. Bioanal. Chem., 401 (2011), pp. 939–955
[SD-008]
[109]
Q. Zang, D.A. Keire, R.D. Wood, L.F. Buhse, C.M. Moore, M. Nasr, A. Al-Hakim, M.L. Trehy, W.J. Welsh
Determination of galactosamine impurities in heparin samples by multivariate regression analysis of their 1H NMR spectra
Anal. Bioanal. Chem., 399 (2011), pp. 635–649
[SD-008]
[110]
V. Ruiz-Calero, J. Saurina, M.T. Galceran, S. Hernández-Cassou, L. Puignou
Potentiality of proton nuclear magnetic resonance and multivariate calibration methods for the determination of dermatan sulfate contamination in heparin samples
Analyst, 125 (2000), pp. 933–938
[SD-008]
[111]
V. Ruiz-Calero, J. Saurina, S. Hernández-Cassou, M.T. Galceran, L. Puignou
Proton nuclear magnetic resonance characterization of glycosaminolgycans using chemometric techniques
Analyst, 127 (2002), pp. 407–415
[SD-008]
[112]
K.R. Riihinen, T. Gödecke, G.F. Pauli
Purification of berry flavonol glycosides by long-bed gel permeation chromatography
J. Chromatogr. A, 1244 (2012), pp. 20–27
[SD-008]
[113]
F. Qiu, B. Friesen, J. McAlpine, G. Pauli
Design of countercurrent separation of Ginkgo biloba terpene lactones by nuclear magnetic resonance
J. Chromatogr. A, 1242 (2012), pp. 26–34
[SD-008]
[114]
A. Tada, K. Takahashi, K. Ishizuki, N. Sugimoto, T. Suematsu, K. Arifuku, M. Tahara, T. Akiyama, Y. Ito, T. Yamazaki
Absolute quantitation of stevioside and rebaudioside A in commercial standards by quantitative NMR
Chem. Pharm. Bull., 61 (2013), pp. 33–38
[SD-008]
[115]
V.G. Dongre, P.D. Ghugare, P. Karmuse, D. Singh, A. Jadhav, A. Kumar
Identification and characterization of process related impurities in chloroquine and hydroxychloroquine by LC/IT/MS, LC/TOF/MS and NMR
J. Pharm. Biomed. Anal., 49 (2009), pp. 873–879
[SD-008]
[116]
B. Raman, B.A. Sharma, R. Butala, P.D. Ghugare, A. Kumar
Structural elucidation of a process-related impurity in ezetimibe by LC/MS/MS and NMR
J. Pharm. Biomed. Anal., 52 (2010), pp. 73–78
[SD-008]
[117]
S. Guntupalli, U. Kumar Raya, N. Muralia, P. Badrinadh Gupta, V.J. Kumar, D. Satheesha, A. Islama
Identification, isolation and characterization of process related impurities in ezetimibe
J. Pharm. Biomed. Anal., 88 (2014), pp. 385–390
[SD-008]
[118]
R. Nageswara Rao, K. Pawan, A. Maurya, Narasa Raju
Isolation and characterization of a potential process related impurity of phenazopyridine HCl by preparative HPLC followed by MS–MS and 2D-NMR spectroscopy
J. Pharm. Biomed. Anal., 49 (2009), pp. 1287–1291
[SD-008]
[119]
D.V.N. Srinivasa Rao, N. Srinivas, Ch. Bharathi, Ch.S. Prasad, A. Ramesh Dandala, Naidu
Identification and characterization of new impurity in didanosine
J. Pharm. Biomed. Anal., 45 (2007), pp. 516–520
[SD-008]
[120]
G. Madhusudhan Reddy, B. Vijaya Bhaskar, P. Pratap Reddy, S. Ashok, P. Sudhakar, J.M. Babu, K. Vyas, K. Mukkanti
Structural identification and characterization of potential impurities of pantoprazole sodium
J. Pharm. Biomed. Anal., 45 (2007), pp. 201–210
[SD-008]
[121]
G. Madhusudhan Reddy, B. Vijaya Bhaskar, P. Pratap Reddy, P. Sudhakar, J. Moses Babu, K. Vyas, P. Ramachandra Reddy, K. Mukkanti
Identification and characterization of potential impurities of rabeprazole sodium
J. Pharm. Biomed. Anal., 43 (2007), pp. 1262–1269
View Record in Scopus
[122]
E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, S. Serra
Impurities of tazarotene: isolation and structural characterization
J. Pharm. Biomed. Anal., 46 (2008), pp. 574–576
Article
|
PDF (180 K)
|
View Record in Scopus
[123]
H. Zhou, Y. Tai, C. Sun, Y. Pan
Separation and characterization of synthetic impurities of triclabendazole by reversed-phase high-performance liquid chromatography/electrospray ionization mass spectrometry
J. Pharm. Biomed. Anal., 37 (2005), pp. 97–107
Article
|
PDF (671 K)
|
View Record in Scopus
[124]
G. Francese, F. Corana, O. Meneghetti, F. Marazza
LC–MS characterization of trace impurities contained in calcium folinate
J. Pharm. Biomed. Anal., 39 (2005), pp. 757–763
Article
|
PDF (437 K)
|
View Record in Scopus
[125]
C. Bharathi, C.S. Prasad, D. Vijaya Bharathi, R. Shankar, V. Janardhana Rao, R. Dandala, A. Naidu
Structural identification and characterization of impurities in ceftizoxime sodium
J. Pharm. Biomed. Anal., 43 (2007), pp. 733–740
Article
|
PDF (489 K)
|
View Record in Scopus
[126]
V. Srinivasan, H. Sivaramakrishnan, B. Karthikeyan
Detection, isolation and characterization of principle synthetic route indicative impurity in telmisartan
Arab. J. Chem. (2012), p. 022 http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.arabjc.2012.03
[127]
A. Sampath, A. Raghupathi Reddy, B. Yakambaram, A. Thirupathi, M. Prabhakar, P. Pratap Reddy, V. Prabhakar Reddy
Identification and characterization of potential impurities of valsartan, AT1 receptor antagonist
J. Pharm. Biomed. Anal., 50 (2009), pp. 405–412
Article
|
PDF (925 K)
|
View Record in Scopus
[128]
B. Raman, B.A. Sharma, G. Mahale, D. Singh, A. Kumar
Investigation and structural elucidation of a process related impurity in candesartan cilexetil by LC/ESI-ITMS and NMR
J. Pharm. Biomed. Anal., 56 (2011), pp. 256–263
Article
|
PDF (421 K)
|
View Record in Scopus
[129]
M. Vuletic, M. Cindric, J.D. Koruznjak
Identification of unknown impurities in simvastatin substance and tablets by liquid chromatography/tandem mass spectrometry
J. Pharm. Biomed. Anal., 37 (2005), pp. 715–721
Article
|
PDF (236 K)
|
View Record in Scopus
[130]
R.M. Bianchini, P.M. Castellano, T.S. Kaufman
Development and validation of an HPLC method for the determination of process-related impurities in pridinol mesylate, employing experimental designs
Anal. Chim. Acta, 654 (2009), pp. 141–147
Article
|
PDF (438 K)
|
View Record in Scopus
[131]
T. Saji, P. Saroj Kumar, J. Subhash Chandra, K. Vineet, A. Ashutosh, V. Dharam
Identification, synthesis and characterization of an unknown process related impurity in eslicarbazepine acetate active pharmaceutical ingredient by LC/ESI-IT/MS, 1H, 13C and 1H-1H COSY NMR
J. Pharm. Anal., 86 (2013), pp. 204–213
View Record in Scopus
[132]
X. Wang, H. Zhou, J. Zheng, C. Huang, W. Liu, L. Yu, S. Zeng
Identification and characterization of four process-related impurities in retigabine
J. Pharm. Biomed. Anal., 71 (2012), pp. 148–151
Article
|
PDF (540 K)
|
View Record in Scopus
[133]
D. Cui, Y. Li, M. Lian, F. Yang, Q. Meng
Development of a simple and stability-indicating RP–HPLC method for determining olanzapine and related impurities generated in the preparative process
Analyst, 136 (2011), pp. 3149–3156
View Record in Scopus
|
Full Text via CrossRef
[134]
G. Goverdhan, A. Raghupathi Reddy, K. Srinivas, V. Himabindu, G. Mahesh Reddy
Identification, characterization and synthesis of impurities of zafirlukast
J. Pharm. Biomed. Anal., 49 (2009), pp. 895–900
Article
|
PDF (531 K)
|
View Record in Scopus
[135]
M. Saravanan, K. Siva kumari, P. Pratap Reddy, M.N. Naidu, J. Moses Babu, A.K. Srivastava, T. Lakshmi Kumar, B.V.V.N. Chandra Sekhar, B. Satyanarayana
Identification, synthesis, isolation and spectral characterization of potential impurities of montelukast sodium
J. Pharm. Biomed. Anal., 48 (2008), pp. 708–715
Article
|
PDF (408 K)
|
View Record in Scopus
[136]
V. Shinde, A. Trivedi, P.R. Upadhayay, N.L. Gupta, D.G. Kanase, R. Chikate
Identification of a new impurity in lisinopril
J. Pharm. Biomed. Anal., 43 (2007), pp. 381–386
Article
|
PDF (279 K)
|
View Record in Scopus
[137]
V.R. Shinde, A. Trivedi, P.R. Upadhayay, N.L. Gupta, D.G. Kanase, R.C. Chikate
Isolation and characterization of benazepril unknown impurity by chromatographic and spectroscopic methods
J. Pharm. Biomed. Anal., 42 (2006), pp. 395–399
Article
|
PDF (252 K)
|
View Record in Scopus
[138]
M. Dousa, P. Gibala, S. Rádl, O. Klecán, Z. Mandelová, J. Brichác
Identification, preparation and UHPLC determination of process-related impurity in zolmitriptan
J. Pharm. Biomed. Anal., 58 (2012), pp. 1–6
Article
|
PDF (170 K)
|
View Record in Scopus
[139]
T.J. Sunder Raj, Ch. Bharathi, M. Saravana Kumar, P. Joseph Prabahar, H. Naveen Kumar, Kumar Sharma, Kalpesh Parikh
Identification, isolation and characterization of process-related impurities in rizatriptan benzoate
J. Pharm. Biomed. Anal., 49 (2009), pp. 156–162
[140]
E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, F. Sala
Isolation and characterisation of impurities in adapalene
J. Pharm. Biomed. Anal., 43 (2007), pp. 1161–1163
Article
|
PDF (140 K)
|
View Record in Scopus
[141]
Y. Liu, L. Chen, Y. Ji
Quantification and structural elucidation of potential impurities in agomelatine active pharmaceutical ingredient
J. Pharm. Biomed. Anal., 81–82 (2013), pp. 193–201
Article
|
PDF (1403 K)
|
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (3)
[142]
B. Raman, B.A. Sharma, P.D. Ghugare, P.P. Karmuse, A. Kumar
Semipreparative isolation and structural elucidation of an impurity in citalopram by LC/MS/MS
J. Pharm. Biomed. Anal., 50 (2009), pp. 377–383
Article
|
PDF (809 K)
|
View Record in Scopus
| |
Citing articles (13)
[143]
B. Raman, B.A. Sharma, P.D. Ghugare, S. Nandavadekar, D. Singh, P.K. Karmuse, A. Kumar
Structural elucidation of process-related impurities in escitalopram by LC/ESI-MS and NMR
J. Pharm. Biomed. Anal., 53 (2010), pp. 895–901
Article
|
PDF (283 K)
|
View Record in Scopus
| |
Citing articles (9)
[144]
Y. Lin, P. Huang, H. Qi, L. Chen, D. Wang
Isolation, synthesis and structure confirmation of the impurity in crude roflumilast product
Res. Chem. Intermed., 39 (2013), pp. 3111–3115
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (2)
[145]
S. Thomas, S. Kumar Paul, S. Shandilya, A. Agarwal, N. Saxena, A. Kumar Awasthi, H.B. Matta, D. Vir, C.S. Mathela
Identification and structural elucidation of two process impurities and stress degradants in darifenacin hydrobromide active pharmaceutical ingredient by LC-ESI/MSn
Analyst, 137 (2012), pp. 3571–3582
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (3)
[146]
P. Elkins, D. Coleman, J. Burgess, M. Gardner, J. Hines, B. Scott, M. Kroenke, J. Larson, M. Lightner, G. Turner, J. White, P. Liu
Characterization of the isomeric configuration and impurities of (Z)-endoxifen by 2D NMR, high resolution LC–MS, and quantitative HPLC analysis
J. Pharm. Biomed. Anal., 88 (2014), pp. 174–179
Article
|
PDF (507 K)
|
View Record in Scopus
| |
Citing articles (3)
[147]
G.R. Bedford, D.N. Richardson
Preparation and identification of cis and trans isomers of a substituted triarylethylene
Nature, 5063 (1966), pp. 733–734
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (41)
[148]
R.P. Shah, A. Sahu, S. Singh
Identification and characterization of geometrical isomeric photo-degradation product of eprosartan using LC–MS and LC–NMR
Eur. J. Chem., 2 (2011), pp. 152–157
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (5)
[149]
M.A. Khan, S. Sinha, S. Vartak, A. Bhartiya, S. Kumar
LC determination of glimepiride and its related impurities
J. Pharm. Biomed. Anal., 39 (2005), pp. 928–943
Article
|
PDF (372 K)
|
View Record in Scopus
| |
Citing articles (31)
[150]
S. Bouabdallah, H. Trabelsi, T. Ben Dhia, S. Sabbah, K. Bouzouita, R. Khaddar
RP–HPLC and NMR study of cis–trans isomerization of enalaprilat
J. Pharm. Biomed. Anal., 31 (2003), pp. 731–741
Article
|
PDF (272 K)
|
View Record in Scopus
| |
Citing articles (16)
[151]
V.G. Dongre, P.P. Karmuse, M.M. Nimbalkar, D. Singh, A. Kumar
Application of GC–EI-MS for the identification and investigation of positional isomer in primaquine, an antimalarial drug
J. Pharm. Biomed. Anal., 39 (2005), pp. 111–116
Article
|
PDF (285 K)
|
View Record in Scopus
| |
Citing articles (15)
[152]
V.G. Dongre, P.P. Karmuse, P.D. Ghugare, M. Gupta, B. Nerurkar, Ch. Shaha, A. Kumar
Characterization and quantitative determination of impurities in piperaquine phosphate by HPLC and LC/MS/MS
J. Pharm. Biomed. Anal., 43 (2007), pp. 186–195
Article
|
PDF (1110 K)
|
View Record in Scopus
| |
Citing articles (10)
[153]
N. Lindegardh, F. Giorgi, B. Galletti, M. Di Mattia, M. Quaglia, D. Carnevale, N.J. White, A. Mazzanti, N.P.J. Day
Identification of an isomer impurity in piperaquine drug substance
J. Chromatogr. A, 1135 (2006), pp. 166–169
Article
|
PDF (243 K)
|
View Record in Scopus
| |
Citing articles (5)
[154]
N. Fukutsu, T. Kawasaki, K. Saito, H. Nakazawa
Application of high-performance liquid chromatography hyphenated techniques for identification of degradation products of cefpodoxime proxetil
J. Chromatogr. A, 1129 (2006), pp. 153–159
Article
|
PDF (473 K)
|
View Record in Scopus
| |
Citing articles (28)
[155]
K.V.V. Prasada Rao, A. Rani, A.V. Raghava Reddy, C.H. Bharathi, R. Dandala, A. Naidu
Isolation, structural elucidation and characterization of impurities in cefdinir
J. Pharm. Biomed. Anal., 43 (2007), pp. 1476–1482
[156]
C.A. Beasley, T.-L. Hwang, K. Fliszar, A. Abenda, D.G. McCollum, R.A. Reed
Identification of impurities in ivermectin bulk material by mass spectrometry and NMR
J. Pharm. Biomed. Anal., 41 (2006), pp. 1124–1134
Article
|
PDF (320 K)
|
View Record in Scopus
| |
Citing articles (4)
[157]
W. Liu, A. Zhou, F. Feng, H. Zhang, J. Zhou, W. Huang
Qualitative and quantitative studies on impurities in G004, a potential hypoglycaemic agent, using liquid chromatography, nuclear magnetic resonance and mass spectrometry
J. Pharm. Biomed. Anal., 56 (2011), pp. 627–632
Article
|
PDF (473 K)
|
View Record in Scopus
| |
Citing articles (4)
[158]
H. Wang, Z. Zhang, F. Xiong, L. Wu, P. Li, W. Ye
Isolation and structure characterization of related impurities in etimicin sulfate by LC/ESI-MSn and NMR
J. Pharm. Biomed. Anal., 55 (2011), pp. 902–907
Article
|
PDF (364 K)
|
View Record in Scopus
| |
Citing articles (6)
[159]
E. Brenna, S. Frigoli, G. Fronza, C. Fuganti, L. Malpezzi
Isolation and characterisation of a phenolic impurity in a commercial sample of duloxetine
J. Pharm. Biomed. Anal., 43 (2007), pp. 1573–1575
Article
|
PDF (131 K)
|
View Record in Scopus
| |
Citing articles (20)
[160]
S.H. Kwak, J.M. Seo, K.-I. Lee
A facile asymmetric synthesis of (S)-duloxetine
ARKIVOC, x (2010), pp. 55–61
View Record in Scopus
| |
Citing articles (1)
[161]
ICH
Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical Substances, Guideline Q6A
International Conference on Harmonisation, IFPMA, Geneva, Switzerland (1999)
[162]
D.W. Armstrong, J.T. Lee, L.W. Chang
Enantiomeric impurities in chiral catalysts, auxiliaries and synthons used in enantioselective synthesis
Tetrahedron: Asymmetry, 9 (1998), pp. 2043–2064
Article
|
PDF (1020 K)
|
View Record in Scopus
| |
Citing articles (33)
[163]
D.W. Armstrong, L.F. He, T. Yu, J.T. Lee, Y.S. Liu
Enantiomeric impurities in chiral catalysts, auxiliaries, synthons and resolving agents. Part 2
Tetrahedron: Asymmetry, 10 (1999), pp. 37–60
Article
|
PDF (1094 K)
|
View Record in Scopus
| |
Citing articles (32)
[164]
K. Huang, Z.S. Breitbach, D.W. Armstrong
Enantiomeric impurities in chiral synthons, catalyst and auxiliaries. Part 3
Tetrahedron: Asymmetry, 17 (2006), pp. 2821–2832
Article
|
PDF (324 K)
|
View Record in Scopus
| |
Citing articles (12)
[165]
R.J. Lewis, M.A. Bernstein, H.-F. Chang, D. Chapman, N. Pemberton
Enantiomeric purity determination by NMR: proving the purity of a single enantiomer
Tetrahedron: Asymmetry, 24 (2013), pp. 866–870
Article
|
PDF (723 K)
|
View Record in Scopus
| |
Citing articles (3)
[166]
T.J. Wenzel
Discrimination of Chiral Compounds Using NMR Spectroscopy
Wiley, Hoboken, USA (2007)
[167]
T.J. Wenzel, C.D. Chisholm
Using NMR spectroscopic methods to determine enantiomeric purity and assign absolute stereochemistry
Prog. Nucl. Magn. Reson. Spectrosc., 59 (2011), pp. 1–63
Article
|
PDF (14543 K)
|
View Record in Scopus
| |
Citing articles (54)
[168]
A. Berthod (Ed.), Chiral Recognition in Separation Methods, Springer, Berlin, Germany (2010)
[169]
G. Tarkanyi
Quantitative approach for the screening of cyclodextrins by nuclear magnetic resonance spectroscopy in support of chiral separations in liquid chromatography and capillary electrophoresis. Enantioseparation of norgestrel with α-, β- and γ-cyclodextrins
J. Chromatogr. A, 961 (2002), pp. 257–276
Article
|
PDF (1306 K)
|
View Record in Scopus
| |
Citing articles (30)
[170]
E. Yashima, C. Yamamoto, Y. Okamoto
NMR studies of chiral discrimination relevant to the liquid chromatographic enantioseparation by a cellulose phenylcarbamate derivative
J. Am. Chem. Soc., 118 (1996), pp. 4036–4048
View Record in Scopus
|
Full Text via CrossRef
| |
Citing articles (155)
[171]
M. Kagawa, Y. Machida, H. Nishi, J. Haginaka
Enantiomeric purity determination of acetyl-l-carnitine by NMR with chiral lanthanide shift reagents
J. Pharm. Biomed. Anal., 38 (2005), pp. 918–923
Article
|
PDF (131 K)
|
View Record in Scopus
| |
Citing articles (8)
[172]
D. Vi Ji, Y.-F. Liu, Q.-H. Wang, C.-F. Zhang, Z.-L. Yang
Isolation, structure characterization and quantification of related impurities in asperosaponin
Chin. J. Nat. Med., 11 (2013), pp. 419–426
[173]
R. Vasu Dev, J. Moses Babu, K. Vyasa, P. Sai Ram, P. Ramachandra, N.M. Sekhar, D.N. Mohan Reddy, N. Srinivasa Rao
Isolation and characterization of impurities in docetaxel
J. Pharm. Biomed. Anal., 40 (2006), pp. 614–622
Article
|
PDF (560 K)
|
View Record in Scopus
| |
Citing articles (26)
[174]
D. Kumar, R. Singh Tomar, S. Kumar Deolia, M. Mitra, R. Mukherjee, A.C. Burman
Isolation and characterization of degradation impurities in docetaxel drug substance and its formulation
J. Pharm. Biomed. Anal., 43 (2007), pp. 1228–1235
Article
|
PDF (250 K)
|
View Record in Scopus
| |
Citing articles (22)
[175]
W. Xu, X. Jia, W. Liu, X. Zhang, Y. Chen, L. Zhou, S. You
Identification and characterization of three impurities in clocortolone pivalate
J. Pharm. Biomed. Anal., 62 (2012), pp. 167–171
Article
|
PDF (485 K)
|
View Record in Scopus
| |
Citing articles (3)
[176]
European Medicines Agency
Committee for Medicinal Products for Human Use, Guideline on the Limits of Genotoxic Impurities, London, UK
(2004)
[177]
C. Bharathi, P. Jayaram, J. Sunder Raj, M. Saravana Kumar, V. Bhargavi, V.K. Handa, R. Dandala, A. Naidu
Identification, isolation and characterization of impurities of clindamycin palmitate hydrochloride
J. Pharm. Biomed. Anal., 48 (2008), pp. 1211–1218
Article
|
PDF (536 K)
|
View Record in Scopus
| |
Citing articles (8)
[178]
Genotoxic Impurities in Pharmaceutical Products Regulations and Analysis, Agilent Technologies, Doc. 5991–1876.
[179]
A. Teasdale (Ed.), Genotoxic impurities: Strategies for Identification and Control, Wiley, New York, USA (2011)
[180]
Validation of quantitative NMR, Implications for early phase of pharmaceutical development, S. de Goeij, MSc. Thesis, Universiteit van Amsterdam, Amsterdam, The Netherlands, 2012.
[181]
I. Raid, Application of qNMR for methanesulfonic acid analysis in pharmaceutical industry, Poster 1180-5. Pittcon 2012, Pittsburgh, USA, March 13, 2012. http://ca.pittcon.org/Technical+Program/tpabstra12.nsf/Agenda+Time+Slots+Web/FC5ADE1BCFBE8CBD852578DA00608AAA?Opendocument (accessed 15.12.13).
[182]
D.P. Elder, D. Snodin, A. Teasdale
Control and analysis of hydrazine, hydrazides and hydrazones—genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products
J. Pharm. Biomed. Anal., 54 (2011), pp. 900–910
Article
|
PDF (272 K)
|
View Record in Scopus
[183]
P. Kovarikova, K. Vavrova, K. Tomalova, M. Schongut, K. Hruskova, P. Hruskova, J. Klimes
HPLC–DAD and MS/MS analysis of novel drug candidates from the group of aromatic hydrazones revealing the presence of geometrical isomers
J. Pharm. Biomed. Anal., 48 (2008), pp. 295–302
Article
|
PDF (273 K)
|
View Record in Scopus
[184]
S. Provera, L. Martini, G. Guercio, L. Turco, L. Costa, C. Marchioro
Application of LC–NMR and HR-NMR to the characterization of biphenyl impurities in the synthetic route development for vestipitant, a novel NK1 antagonist
J. Pharm. Biomed. Anal., 53 (2010), pp. 389–395
Article
|
PDF (315 K)
|
View Record in Scopus
[185]
K.W. Sigvardson, S.P. Adams, T. Bradford Barnes, K.F. Blom, J.M. Fortunak, M.J. Haas, K.L. Reilly, A.J. Repta, G.A. Nemeth
The isolation and identification of a toxic impurity in XP315 drug substance
J. Pharm. Biomed. Anal., 27 (2002), pp. 327–334
Article
|
PDF (154 K)
|
View Record in Scopus
[186]
I.C.H.
Impurities Guideline for residual solvents Q3C(R5)
International Conference on Harmonisation, IFPMA, Geneva, Switzerland (2011)
[187]
C. B’Hymer
Residual solvent testing: a review of gas-chromatographic and alternative techniques
Pharm. Res., 20 (2003), pp. 337–343
[188]
M. Guerrini, A. Bisio, G. Torri
Combined quantitative 1H and 13C nuclear magnetic resonance spectroscopy for characterization of heparin preparations
Semin. Thromb. Hemost., 27 (2001), pp. 473–482
View Record in Scopus
|
Full Text via CrossRef
[189]
F. Zhang, B. Yang, M. Ly, K. Solakyildirim, Z. Xiao, Z. Wang, J.M. Beaudet, A.Y. Torelli, J.S. Dordick, R.J. Linhardt
Structural characterization of heparins from different commercial sources
Anal. Bioanal. Chem., 401 (2011), pp. 2793–2803
View Record in Scopus
|
Full Text via CrossRef
[190]
I. McEwen, A. Amini, I.M. Olofsson
Identification and purity test of heparin by NMR. A summary of 2 years’ experience at the medical products agency
Pharmaeuropa Bio Sci. Notes, 1 (2010), pp. 65–72
View Record in Scopus
[191]
G.A. Neville, F. Mori, K.R. Holme, A.S. Perlin
Monitoring the purity of pharmaceutical heparin preparations by high-field 1HNMR magnetic resonance spectroscopy
J. Pharm. Sci., 78 (1989), pp. 101–104
View Record in Scopus
|
Full Text via CrossRef
[192]
M.D. Argentine, P.K. Owens, B.A. Olsen
Strategies for the investigation and control of process-related impurities in drug substances
Adv. Drug Deliv. Rev., 59 (2007), pp. 12–28
Article
|
PDF (841 K)
|
View Record in Scopus
[193]
P.F. Bross, R. Kane, A.T. Farrell, S. Abraham, K. Benson, M.E. Brower, S. Bradley, J.V. Gobburu, A. Goheer, S.L. Lee, J. Leighton, C.Y. Liang, R.T. Lostritto, W.D. McGuinn, D.E. Morse, A. Rahman, L.A. Rosario, S.L. Verbois, G. Williams, Y.C. Wang, R. Pazdur
Approval summary for bortezomib for injection in the treatment of multiple myeloma
Clin. Cancer Res., 10 (2004), pp. 3954–3964
View Record in Scopus
|
Full Text via CrossRef
[194]
S.R. Byrn, P.A. Tishmack, M.J. Milton, H. van de Velde
Analysis of two commercially available bortezomib products: differences in assay of active agent and impurity profile
AAPS PharmSciTech, 12 (2011), pp. 461–467
View Record in Scopus
|
Full Text via CrossRef
[195]
J. Forshed, B. Erlandsson, S.P. Jacobsson
Quantification of aldehyde impurities in poloxamer by 1H NMR spectrometry
Anal. Chim. Acta, 552 (2005), pp. 160–165
Article
|
PDF (236 K)
|
View Record in Scopus
[196]
C. Thomasin, P. Johansen, R. Alder, R. Besmsel, G. Hottinger, H. Altorfer, A.D. Wright, G. Wehrli, H.P. Merkle, B. Gander
A contribution to overcoming the problems of residual solvents in biodegradable microspheres prepared by coacervation
J. Pharm. Biopharm., 42 (1996), pp. 16–24
View Record in Scopus
[197]
H.W. Avdovich, M.J. Lebelle, C. Savard, W.L. Wilson
Nuclear magnetic resonance identification of solvent residue in cocaine
Forensic Sci. Int., 49 (1991), pp. 225–235
Article
|
PDF (690 K)
|
View Record in Scopus
[198]
R.J. Mumper, M. Jay
Poly(l-lactic acid) microspheres containing neutron-activatable holmium-165: a study of the physical characteristics of microspheres before and after irradiation in a nuclear reactor
Pharm. Res., 9 (1992), pp. 149–154
View Record in Scopus
|
Full Text via CrossRef
[199]
D. Jain, P.K. Basniwal
A forced degradation and impurity profiling: recent trends in analytical perspectives
J. Pharm. Biomed. Anal., 86 (2013), pp. 11–35
Article
|
PDF (1423 K)
|
View Record in Scopus
[200]
S. Singh, M. Junwal, G. Modhe, H. Tiwari, M. Kurmi, N. Parashar, P. Sidduri
Forced degradation studies to assess the stability of drugs and products
Trends Anal. Chem., 49 (2013), pp. 71–88
Article
|
PDF (520 K)
|
View Record in Scopus
[201]
A. Awasthi, M. Razzak, R. Al-Kassas, D.R. Greenwood, J. Harvey, S. Garg
Separation and identification of degradation products in eprinomectin formulation using LC, LTQ FT-MS, H/D exchange, and NMR
J. Pharm. Biomed. Anal., 63 (2012), pp. 62–73
Article
|
PDF (1484 K)
|
View Record in Scopus
[202]
R.N. Rao, K. Ramakrishna, B. Sravan, K. Santhakumar
RP–HPLC separation and ESI-MS, 1H, and 13C NMR characterization of forced degradants including process related impurities of carisbamate: method development and validation
J. Pharm. Biomed. Anal., 77 (2013), pp. 49–54
[SD-008]
[203]
A. Mohan, M. Hariharan, E. Vikraman, G. Subbaiah, B.R. Venkataraman, D. Saravanan
Identification and characterization of a principal oxidation impurity in clopidogrel drug substance and drug product
J. Pharm. Biomed. Anal., 47 (2008), pp. 183–189
[SD-008]
[204]
R.M. Bianchini, P.M. Castellano, T.S. Kaufman
Validated stability-indicating HPLC method for the determination of pridinol mesylate. Kinetics study of its degradation in acid medium
J. Pharm. Biomed. Anal., 48 (2008), pp. 1151–1160
[SD-008]
[205]
S. Doddaga, R. Peddakonda
Chloroquine-N-oxide, a major oxidative degradation product of chloroquine: identification, synthesis and characterization
J. Pharm. Biomed. Anal., 81–82 (2013), pp. 118–125
[SD-008]
[206]
T. Dyakonov, A. Muir, H. Nasri, D. Toops, A. Fatmi
Isolation and characterization of cetirizine degradation product: mechanism of cetirizine oxidation
Pharm. Res., 27 (2010), pp. 1318–1324
[SD-008]
[207]
C. Ashok, M. Golak, D. Adwait, V. Krishna, A. Himani, P. Umesh, G. Amol, S. Srinivas, M. Sharad, J. Deepali, C. Atul
Isolation and structural elucidation of two impurities from a diacerein bulk drug
J. Pharm. Biomed. Anal., 49 (2009), pp. 525–528
[SD-008]
[208]
B.V. Subbaiah, K.K.S. Ganesh, G.V. Krishna, K. Vyas, R.V. Dev, K.S. Reddy
Isolation and characterisation of degradant impurities in dipyridamole formulation
J. Pharm. Biomed. Anal., 61 (2012), pp. 256–264
[SD-008]
[209]
P. Novak, P. Tepes, M. Ilijas, I. Fistric, I. Bratos, A. Avdagic, Z. Hameršak, V.G. Marković, M. Dumić
LC–NMR and LC–MS identification of an impurity in a novel antifungal drug icofungipen
J. Pharm. Biomed. Anal., 50 (2009), pp. 68–72
[SD-008]
[210]
C. Sun, J. Wu, D. Wang, Y. Pan
Characterization of a novel impurity in bulk drug eprosartan by ESI/MSn and NMR
J. Pharm. Biomed. Anal., 51 (2010), pp. 778–783
[SD-008]
[211]
A. Mohan, S. Shanmugavel, A. Goyal, B.R. Venkataraman, D. Saravanan
Identification, isolation, and characterization of five potential degradation impurities in candesartan cilexetil tablets
Chromatographia, 69 (2009), pp. 1211–1220
[SD-008]
[212]
S. Provera, L. Rovatti, L. Turco, S. Mozzo, A. Spezzaferri, S. Bacchi, A. Ribecai, S. Guelfi, A. Mingardi, C. Marchioro, D. Papini
A multi-technique approach using LC–NMR, LC–MS, semi-preparative HPLC, HR–NMR and HR–MS for the isolation and characterization of low-level unknown impurities in GW876008, a novel corticotrophin-release factor 1 antagonist
J. Pharm. Biomed. Anal., 53 (2010), pp. 517–525
[SD-008]
[213]
C. Sitaram, R. Rupakula, B.N. Reddy
Determination and characterization of degradation products of anastrozole by LC–MS/MS and NMR spectroscopy
J. Pharm. Biomed. Anal., 56 (2011), pp. 962–968
[SD-008]
[214]
W. Feng, H. Liu, G. Chen, R. Malchow, F. Bennett, E. Lin, B. Pramanik, T.M. Chan
Structural characterization of the oxidative degradation products of an anti-fungal agent SCH 56592 by LC–NMR and LC–MS
J. Pharm. Biomed. Anal., 25 (2001), pp. 545–557
[SD-008]
[215]
A. Awasthi, M. Razzak, R. Al-Kassas, D.R. Greenwood, J.H. Sanjay Garg
Isolation and characterization of degradation products of moxidectin using LC, LTQ FT-MS, H/D exchange and NMR
Anal. Bioanal. Chem., 404 (2012), pp. 2203–2222
[SD-008]
[216]
J. Wang, W. Li, C.-G. Li, Y.-Z. Hu
Photodegradation of fleroxacin injection: different products with different concentration levels
AAPS PharmSciTech, 12 (2011), pp. 872–878
[SD-008]
[217]
A. Kugimiya, H. Naoki, N. Hamanaka, E. Nishimura
Photo-degradation products of pramipexole
Bioorg. Med. Chem. Lett., 22 (2012), pp. 2951–2953
[SD-008]
[218]
R.P. Shah, S. Singh
Identification and characterization of a photolytic degradation product of telmisartan using LC-MS/TOF, LC-MSn, LC–NMR and on-line H/D exchange mass studies
J. Pharm. Biomed. Anal., 53 (2010), pp. 755–761
[SD-008]
[219]
R.P. Shah, A. Sahu, S. Singh
Identification and characterization of degradation products of irbesartan using LC–MS/TOF, MSn, on-line H/D exchange and LC–NMR
J. Pharm. Biomed. Anal., 51 (2010), pp. 1037–1046
[SD-008]
[220]
A.K. Gajjar, V.D. Shah
Isolation and structure elucidation of major alkaline degradant of ezetimibe
J. Pharm. Biomed. Anal., 55 (2011), pp. 225–229
[SD-008]
[221]
C.R. Barhate, K. Mohanraj
What is the degradation product of ezetimibe?
J. Pharm. Biomed. Anal., 55 (2011), pp. 1237–1238
Article
|
PDF (121 K)
|
View Record in Scopus
|
Citing articles (5)
[222]
Z. Sánta, J. Koti, K. Szőke, K. Vukics, C. Szántay Jr.
Structure of the major degradant of ezetimibe
J. Pharm. Biomed. Anal., 58 (2012), pp. 125–129
Article
|
PDF (176 K)
|
View Record in Scopus
|
Citing articles (4)
[223]
G.Y.S.K. Swamy, K. Ravikumar, L.K. Wadhwa, R. Saxena, S. Singh
(2R* 3R*,6S*)-N,6-Bis(4-fluorophenyl)-2-(4-hydroxyphenyl)-3,4,5,6-tetrahydro-2H-pyran-3-carboxamide
Acta Crystallogr. E, 61 (2005), pp. o3608–o3610
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (9)
[224]
K. Filip, K. Bankowski, K. Sidoryk, J. Zagrodzka, M. Łaszcz, K. Trzcinska, A. Szyprowska, P. Cmoch, W. Maruszak
Physicochemical characterization of ezetimibe and its impurities
J. Mol. Struct., 991 (2011), pp. 162–170
Article
|
PDF (1463 K)
|
View Record in Scopus
|
Citing articles (9)
[225]
A. Mendez, P. Chagastelles, E. Palma, N. Nardi, E. Schapoval
Thermal and alkaline stability of meropenem: degradation products and cytotoxicity
Int. J. Pharm., 350 (2008), pp. 95–102
Article
|
PDF (407 K)
|
View Record in Scopus
|
Citing articles (27)
[226]
R. Nageswara Rao, A. Narasa Raju, R. Narsimha
Isolation and characterization of process related impurities and degradation products of bicalutamide and development of RP–HPLC method for impurity profile study
J. Pharm. Biomed. Anal., 46 (2008), pp. 505–519
Article
|
PDF (1514 K)
|
View Record in Scopus
|
Citing articles (19)
[227]
R.N. Rao, Ch., G. Naidu, K.G. Prasad, B. Santhakumar, S. Saida
Development and validation of a stability indicating assay of doxofylline by RP–HPLC: ESI–MS/MS, 1H and 13C NMR spectroscopic characterization of degradation products and process related impurities
J. Pharm. Biomed. Anal., 78–79 (2013), pp. 92–99
Article
|
PDF (965 K)
|
View Record in Scopus
|
Citing articles (4)
[228]
A.J. Lin, L.Q. Li, D.L. Klayman, C.F. George, J.L. Flippen-Anderson
Antimalarial activity of new water-soluble dihydroartemisinin derivatives. 3. Aromatic amine analogs
J. Med. Chem., 33 (1990), pp. 2610–2614
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (56)
[229]
C. Charrier, G. Bertho, O. Petigny, P. Moneton, R. Azerad
A new derivative detected in accelerated ageing of artesunate-amodiaquine fixed dose combination tablets
J. Pharm. Biomed. Anal., 81–82 (2013), pp. 20–26
Article
|
PDF (977 K)
|
View Record in Scopus
|
Citing articles (1)
[230]
M. Xia, T.-J. Hang, F. Zhang, X.-M. Li, X.-Y. Xu
The stability of biapenem and structural identification of impurities in aqueous solution
J. Pharm. Biomed. Anal., 49 (2009), pp. 937–944
Article
|
PDF (993 K)
|
View Record in Scopus
|
Citing articles (21)
[231]
Z. Béni, V. Háda, E. Varga, S. Mahó, A. Aranyi, C. Szántay Jr.
New oxidative decomposition mechanism of estradiol through the structural characterization of a minute impurity and its degradants
J. Pharm. Biomed. Anal., 78–79 (2013), pp. 183–189
Article
|
PDF (377 K)
|
View Record in Scopus
|
Citing articles (2)
[232]
T. Murakami, H. Konno, N. Fukutsu, M. Onodera, T. Kawasaki, F. Kusu
Identification of a degradation product in stressed tablets of olmesartan medoxomil by the complementary use of HPLC hyphenated techniques
J. Pharm. Biomed. Anal., 47 (2008), pp. 553–559
Article
|
PDF (821 K)
|
View Record in Scopus
|
Citing articles (35)
[233]
E. Del Grosso, S. Aprile, G. Grosa
Forced degradation study of thiocolchicoside: characterization of its degradation products
J. Pharm. Biomed. Anal., 61 (2012), pp. 215–223
[234]
R. Azerad
Microbial transformations of artemisinin and artemisinin derivatives: an example of the microbial generation of molecular diversity
Curr. Bioact. Compd., 8 (2012), pp. 151–158
Full Text via CrossRef
[235]
W.-M. Wu, Y. Wu, Y.-L. Wu, Z.-J. Yao, C.-M. Zhou, Y. Li, F. Shan
Unified mechanistic framework for the Fe(II)-induced cleavage of Qinghaosu and derivatives/analogues
The first spin-trapping evidence for the previously postulated secondary C-4 radical, J. Am. Chem. Soc., 120 (1998), pp. 3316–3325
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (150)
[236]
R.K. Haynes, W.C. Chan, C.-M. Lung, A.-C. Uhlemann, U. Eckstein, D. Taramelli, S. Parapini, D. Monti, S. Krishna
The Fe2+-mediated decomposition, PfATP6 binding, and antimalarial activities of artemisone and other artemisinins: the unlikelihood of C-centered radicals as bioactive intermediates
Chem. Med. Chem., 2 (2007), pp. 1480–1497
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (60)
[237]
C.D. Sommers, E.S. Pang, H. Ghasriani, R.T. Berendt, V.L. Vilker, D.A. Keire, M.T. Boyne
II. Analyses of marketplace tacrolimus drug product quality: bioactivity, NMR and LC–MS
J. Pharm. Biomed. Anal., 85 (2013), pp. 108–117
Article
|
PDF (1109 K)
|
View Record in Scopus
|
Citing articles (1)
[238]
P. Ferraboschi, D. Colombo, M. de Mieri, P. Grisenti
Evaluation, synthesis and characterization of tacrolimus impurities
J. Antibiotics, 65 (2012), pp. 349–354
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (3)
[239]
A. Subasranjan, R. Hemant
An improved validated ultra-high pressure liquid chromatography method for separation of tacrolimus impurities and its tautomers
Drug Test. Anal., 2 (2010), pp. 107–112
View Record in Scopus
|
Citing articles (5)
[240]
D.F. Mierke, P. Schmieder, P. Karuso, H. Kessler
Conformational analysis of the cis- and trans-isomers of FK506 by NMR and molecular dynamics
Helv. Chim. Acta, 74 (1991), pp. 1027–1047
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (21)
[241]
B.E. Segmuller, B.L. Armstrong, R. Dunphy, A.R. Oyler
Identification of autoxidation and photodegradation products of ethynylestradiol by on-line HPLC–NMR and HPLC–MS
J. Pharm. Biomed. Anal., 23 (2000), pp. 927–937
Article
|
PDF (179 K)
|
View Record in Scopus
|
Citing articles (37)
[242]
M. Pendela, S. Béni, E. Haghedooren, L. Van den Bossche, B. Noszál, A. Van Schepdael, J. Hoogmartens, E. Adams
Combined use of liquid chromatography with mass spectrometry and nuclear magnetic resonance for the identification of degradation compounds in an erythromycin formulation
Anal. Bioanal. Chem., 402 (2012), pp. 781–790
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (7)
[243]
C. Pan, J. Guan, M. Lin
A multidisciplinary approach to identify a degradation product in a pharmaceutical dosage form
J. Pharm. Biomed. Anal., 54 (2011), pp. 855–859
Article
|
PDF (284 K)
|
View Record in Scopus
|
Citing articles (5)
[244]
C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
Determination of free fatty acids in pharmaceutical lipids by 1H NMR and comparison with the classical acid value
J. Pharm. Biomed. Anal. (2013) http://dx.doi.org.ezaccess.library.uitm.edu.my/10.1016/j.jpba.04.010
[245]
C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
Determination of free fatty acids in edible oils by 1H NMR spectroscopy
Lipid Technol., 24 (2012), pp. 279–281
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (7)
[246]
C. Skiera, P. Steliopoulos, T. Kuballa, B. Diehl, U. Holzgrabe
1H NMR spectroscopy as a new tool in the assessment of the oxidative state in edible oils
J. Am. Oil Chem. Soc., 89 (2012), pp. 1383–1391
View Record in Scopus
|
Citing articles (9)
[247]
H. Böhme, K. Hartke, H. Hartke, M. Wichtl
Kommentar zum Europäischen Arzneibuch, Vol. 41, Aktualisierungslieferung
Wissenschaftliche Verlagsge-sellschaft, Stuttgart, Germany (2012)
[248]
S. Krist, G. Buchbauer, C. Klausberger
Lexikon der pflanzlichen Öle und Fette
Springer, Vienna, Austria (2008)
[249]
R.A. Seburg, J.M. Ballard, T.-L. Hwang, C.M. Sullivan
Photosensitized degradation of losartan potassium in an extemporaneous suspension formulation
J. Pharm. Biomed. Anal., 42 (2006), pp. 411–422
Article
|
PDF (471 K)
|
View Record in Scopus
|
Citing articles (24)
[250]
T.-M. Chan, B. Pramanik, R. Aslanian, V. Gullo, M. Patel, B. Cronin, C. Boyce, K. McCormick, M. Berlin, X. Zhu, A. Buevich, L. Heimark, P. Bartner, G. Chen, H. Pu, V. Hegd
Isolation, structural determination, synthesis and quantitative determination of impurities in Intron-A, leached from a silicone tubing
J. Pharm. Biomed. Anal., 49 (2009), pp. 327–332
Article
|
PDF (391 K)
|
View Record in Scopus
|
Citing articles (3)
[251]
J. Larsen, D. Staerk, C. Cornett, S.H. Hansen, J.W. Jaroszewski
Identification of reaction products between drug substances and excipients by HPLC–SPE–NMR: ester and amide formation between citric acid and 5-aminosalicylic acid
J. Pharm. Biomed. Anal., 49 (2009), pp. 839–842
Article
|
PDF (269 K)
|
View Record in Scopus
|
Citing articles (15)
[252]
P. Novak, P. Tepes, I. Fistric, I. Bratos, V. Gabelica
The application of LC–NMR and LC-MS for the separation and rapid structure elucidation of an unknown impurity in 5-aminosalicylic acid
J. Pharm. Biomed. Anal., 40 (2006), pp. 1268–1272
Article
|
PDF (185 K)
|
View Record in Scopus
|
Citing articles (22)
[253]
M. Dousa, P. Gibala, J. Havlicek, L. Placek, M. Tkadlecova, J. Bricha
Drug-excipient compatibility testing–Identification and characterization of degradation products of phenylephrine in several pharmaceutical formulations against the common cold
J. Pharm. Biomed. Anal., 55 (2011), pp. 949–956
Article
|
PDF (584 K)
|
View Record in Scopus
|
Citing articles (4)
[254]
A. Marín, A. Espada, P. Vidal, C. Barbas
Major degradation product identified in several pharmaceutical formulations against the common cold
Anal. Chem., 77 (2005), pp. 471–477
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (17)
[255]
O. Galanopoulou, S. Rozou, E. Antoniadou-Vyza
HPLC analysis, isolation and identification of a new degradation product in carvedilol tablets
J. Pharm. Biomed. Anal., 48 (2008), pp. 70–77
Article
|
PDF (692 K)
|
View Record in Scopus
|
Citing articles (14)
[256]
J. Battiste, R.A. Newmark
Applications of 19F multidimensional NMR
Prog. Nucl. Magn. Reson. Spectrosc., 48 (2006), pp. 1–23
Article
|
PDF (1112 K)
|
View Record in Scopus
|
Citing articles (46)
[257]
S. Trefi, V. Gilard, M. Malet-Martino, R. Martino
Generic ciprofloxacin tablets contain the stated amount of drug and different impurity profiles: a19F, 1H and DOSY NMR analysis
J. Pharm. Biomed. Anal., 44 (2007), pp. 743–754
Article
|
PDF (689 K)
|
View Record in Scopus
|
Citing articles (13)
[258]
S. Trefi, V. Gilard, S. Balayssac, M. Malet-Martino, R. Martino
Quality assessment of fluoxetine and fluvoxamine pharmaceutical formulations purchased in different countries or via the Internet by 19F and 2D DOSY 1H NMR
J. Pharm. Biomed. Anal., 46 (2008), pp. 707–722
Article
|
|
Citing articles (20)
[259]
V.G. Dongre, P.D. Ghugare, P.P. Karmusea, S.R. Soudagar, N. Panda, A. Kumar
Isolation and structural identification of an impurity in fluconazole bulk drug substance
J. Pharm. Biomed. Anal., 45 (2007), pp. 422–429
Article
|
PDF (909 K)
|
View Record in Scopus
|
Citing articles (9)
[260]
B. Marciniec, K. Dettlaff, E. Jaroszkiewicz, J. Bafeltowska
Radiochemical stability of fluconazole in the solid state
J. Pharm. Biomed. Anal., 43 (2007), pp. 1876–1880
Article
|
PDF (471 K)
|
View Record in Scopus
|
Citing articles (13)
[261]
L. Wu, T.Y. Hong, F.G. Vogt
Structural analysis of photo-degradation in thiazole-containing compounds by LC–MS/MS and NMR
J. Pharm. Biomed. Anal., 44 (2007), pp. 763–772
Article
|
PDF (644 K)
|
View Record in Scopus
|
Citing articles (12)
[262]
J. Brusa, M. Urbanova, I. Sedenkova, H. Brusova
New perspectives of 19F MAS NMR in the characterization of amorphous forms of atorvastatin in dosage formulations
Int. J. Pharm., 409 (2011), pp. 62–74
[263]
M. Nisha, M. Ismail, R. Ismail, F. Duncan, L. Maili, K.N. Jeremy, C.L. John
Impurity profiling in bulk pharmaceutical batches using 19F NMR spectroscopy and distinction between monomeric and dimeric impurities by NMR-based diffusion measurements
J. Pharm. Biomed. Anal., 19 (1999), pp. 511–517
[264]
J.L. Battiste, N. Jing, R.A. Newmark
2D 19F–19F NOESY for the assignment of NMR spectra of fluorochemicals
J. Fluorine Chem., 125 (2004), pp. 1331–1337
Article
|
PDF (321 K)
|
View Record in Scopus
|
Citing articles (26)
[265]
W. Zhong, B. Hilton, G. Martin, L. Wang, S.H. Yip
Identification of a unique cationic impurity in Preladenant™ using accurate MS and NMR
J. Heterocycl. Chem., 50 (2013), pp. 281–287
View Record in Scopus
|
Full Text via CrossRef
|
Citing articles (1)
[266]
R.M. Bianchini, P.M. Castellano, T.S. Kaufman
Characterization of two new potential impurities of valsartan obtained under photodegradation stress condition
J. Pharm. Biomed. Anal., 56 (2011), pp. 16–22


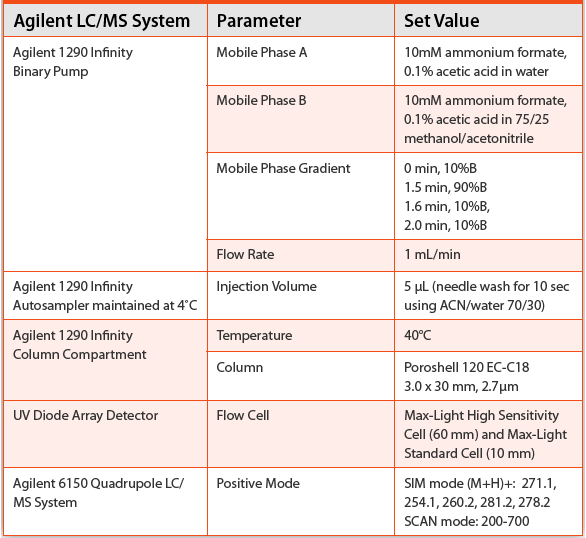

{kind=link}