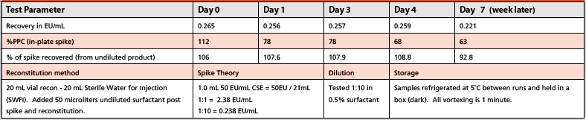
Abstract
There is considerable interest in enhancing approaches for analytical method development, validation, and lifecycle management which are aligned with Quality by Design (QbD) approaches to drug development. These enhanced approaches utilize risk assessments and in-depth systematic evaluations of critical method variables to identify those controls required to ensure that the measurement uncertainty of a reportable result is controlled to a level that ensures that the method is “fit for purpose” (ie, suitable for its intended original use). Predefined criteria can be established in the form of an Analytical Target Profile (ATP), which contains the desired performance criteria for the measurement of the attribute(s), eg, impurities content, dissolution, potency, etc. The ATP defines the appropriate performance criteria for the analytical result and is method-independent.
In a QbD approach, the intended product control strategy needs to be defined in order to determine what analytical methods are required and where in the process they need to be employed. Toxicological-based safety limits (staged Threshold of Toxicological Concern, Acceptable Daily Intake, etc), links to clinical efficacy, and process performance across the product design space should then be used to determine the specification limits and required ATPs for all the measured attributes. This differs from the traditional approach of developing end-product tests based on batch data which may be restricted to operation across a limited region of the design space. During regulatory review, it is common for proposed specification limits to be challenged—and subsequently narrowed—on the basis of manufacturing batch experience aligned with traditional concepts of specification setting outlined in ICHQ6. This may have major consequences for the control strategy including the method(s) no longer being fit for purpose. This critical interplay between specification limits and the underpinning methods is often not fully understood.
Introduction
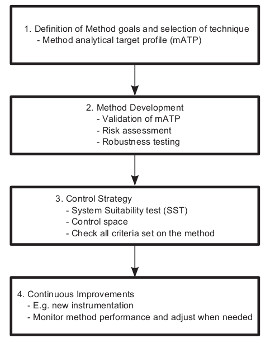
There is considerable interest in enhancing approaches for analytical method development, validation, and lifecycle management which are aligned with Quality by Design (QbD) approaches to drug development. The application of QbD to analytical methods (also known as analytical procedures or test procedures) was initially proposed by Borman, et al.1 Following this publication, the Pharmaceutical Research and Manufacturers of America (PhRMA) and European Federation of Pharmaceutical Industries Association (EFPIA) set up the Analytical Technical Group (ATG) and the Analytical Design Space (ADS) topic teams, respectively, to progress across the industry how QbD could be applied to the lifecycle of analytical methods. In 2010, these teams published a joint white paper2 defining key elements and benefits for the application of enhanced analytical life-cycle approaches (Figure 1) in alignment with ICH chapters Q8, Q9, and Q10. Figure 1. Components of application of QbD to analytical methods as defined by the PhRMA ATG and EFPIA ADS Teams.2
Figure 1. Components of application of QbD to analytical methods as defined by the PhRMA ATG and EFPIA ADS Teams.2
Enhanced approaches during the “Method Evaluation” phase were proposed which utilized risk assessments and in-depth systematic evaluations (eg, robustness and ruggedness testing3-4) of critical method variables to identify those controls required to ensure that the measurement uncertainty of a reportable result is controlled to a level that ensures that the method is “fit for purpose” (ie, suitable for its intended original use). The new concept of the Analytical Target Profile (ATP) analogous to the Quality Target Product Profile5 (QTPP) was introduced which was described as “the method requirements needed to adequately measure the defined CQAs (critical quality attributes) of the Drug Product.”2 More recently, the U.S. Pharmacopeial (USP) Validation and Verification Expert Panel have further defined the ATP as “the objective of the test and quality requirements, including the expected level of confidence, for the reportable result that allows the correct conclusion to be drawn regarding the attributes of the material that is being measured.”6 The European Medicines Agency and U.S. Food and Drug Administration have also stated that the ATP “can be acceptable as a qualifier of the expected method performance by analogy to the QTPP.”7 Dialogue, however, needs to continue between regulators and industry to establish a globally harmonized definition of the ATP.
It is, therefore, the ATP that should drive the design, development, and validation of appropriate analytical methods. However, this presents challenges during drug development. The intended product control strategy needs to be defined in order to determine what analytical methods are required and where in the process they need to be employed. The control strategy is often not fully defined until quite late in the development cycle, which often results in the need for a generic ATP—in early development—related to the expected performance of an analytical technique. This early-development, generic ATP then transitions into the commercial ATP once the control strategy is finalized. If a design space is being used as part of the control strategy, then the ATP must cover the control requirements across the entirety of the design space. Finally, specification limits are often changed in late phase development for several reasons, including changes in regulatory guidance or as a result of gaining more process understanding. Late changes to specification limits may result in the ATP no longer being met, and the method(s) no longer being fit for purpose.
Attribute Controls within the Control Strategy
A CQA is defined as “a physical, chemical, biological or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality.”5 Hence, establishing the link between an attribute and the safety and efficacy of a product is key to identifying which of those attributes are CQAs and should be the focus of the control strategy.
A control strategy can include, but is not limited to, the following5,8,9:
- Attribute controls on material attributes (including raw materials, starting materials, intermediates, excipients, reagents, primary packaging materials, etc)
- Controls implicit in the design of the manufacturing process (eg, sequence of purification steps; use of “delumping” processes; order of addition of reagents)
- In-process attribute controls (eg, in-process tests, process analytical technology)
- In-process parametric controls (eg, control of critical process parameters)
- Controls on a drug substance and drug product (eg, release testing)
- GMP controls
In the first instance, consideration of the QTPP will identify many of the CQAs. For example, control of CQAs such as content uniformity and dissolution is essential to ensure the efficacy of solid oral dosage forms, whereas aerosolized particle size distribution will typically be a CQA for an inhaled formulation.
Other attributes may require further assessment, potentially including experimental studies, to determine whether there is a potential impact on the product quality. For example, only those attributes of the active pharmaceutical ingredient (API) which impact product quality are CQAs.8 Hence, in some cases, API particle size may impact dissolution of a tablet or capsule, but in others (eg, where the API is BCS class 1) this may not be the case. Equally, other API physical properties such as habit or particle surface roughness may impact quality in other formulations. A further example is that there may be potential drug-related impurities or solvents used in the API manufacturing process which are very efficiently purged downstream, therefore, posing no risk to quality. Such attributes need not be considered CQAs and their control strategy can be aligned to this low level of risk. Equally, genotoxic (mutagenic) impurities, for which very low levels present a significant risk, could all be considered as API CQAs and the control strategy suitably developed.9
In many cases, the overall control strategy may include controls at several points in the manufacturing process (see Figure 2 for an excerpt from a control strategy). Ideally, a control strategy will endeavor to ensure control of CQAs as early in the manufacturing process as possible. Hence, establishing the input raw material specification as the primary control point of any impurity in that raw material, which may track through or be transformed into a CQA impurity, is preferable to testing only at an intermediate stage or the final API. Zoom In
Zoom In
Figure 2. A visual summary of part of a control strategy for an API process, highlighting the main control points in API, the raw materials, and the manufacturing steps.
A fundamental consideration in developing each control element is the role it plays in the overall control strategy. Control elements, such as the reduction or purging of impurities under certain processing conditions, can often be coupled with additional controls in the raw materials and end products or intermediates. It is possible that any one of these control points alone provides a control strategy appropriate for the quality of the product and hence it may be argued on the basis of scientific understanding and risk that additional points of control are not required.
However, no single control element is 100% risk-free: for an in-process or end product test, method reliability (eg, method variability or potential sources of bias) and the importance of the process control are key considerations in determining the risk which will inform the enhanced method validation activities. Equally, for a CQA controlled by a design space (ie, the combination of process variables such as parameters and raw material attributes), the risk associated with control by the design space alone will vary as a function of the design space model fit and process variation, its applicability to the scale and equipment selected, and the proximity of the defined process boundaries to the edge of failure.
The risk associated with a control strategy including both the design space and the end-product testing will be reduced when both are used in combination. Hence, consideration of the overall control strategy should be a key factor that is considered when the individual control elements are developed.
Specification Setting in a Design Space World
Specifications are key elements of the control strategy. Alignment of specification setting with QbD principles will provide greater assurance of product safety.
Previously, specifications were set utilizing mainly end product testing and with the limits aligned to ensure safety, clinical efficacy, product quality, and manufacturing consistency. Limits were set based on historical batch data from the proposed process (route, process and scale) and taking into account acceptable limits set out in ICH Q3A,10 Q3B,11 Q6A,12 and M7.9 Specification limits have often been unnecessarily tight because they have been based on a limited number of batches all operated at or close to the target parameter setpoints. (target-point batches). Figure 3 illustrates how specifications should relate to safety, efficacy, and the process design space using an impurity as an example. Figure 3. Impurity Specification: Illustration of the relationship between patient safety, efficacy, and the design space.
Figure 3. Impurity Specification: Illustration of the relationship between patient safety, efficacy, and the design space.
In a design space world, the setting of specifications and the definition of specification limits will be based on:
- Safety qualification, toxicological-based safety limits and ICH Guidance9,10,11,13
- Clinical efficacy
- Process understanding and performance (Design Space Evaluation, ICH Q8,5 Q118)
- Review of target-point batches to ensure specifications are met and process laboratory models are verified at scale (compared against the predicted mean and expected variability)
- More reliance on input and process control, in-process monitoring and testing, and less dependence on end product testing
Specifications will become more science-based. Specification limits must be based on the manufacturing process understanding for the process operated across the defined design space. Target-point batch data alone is no longer suitable. Data and predictive models presented across the whole design space are likely to lead to wider specification limits so that the safety and efficacy of the product are maintained across the design space. Tighter limits relating to where the process is being operated in the design space can be applied within a company using statistical process control14 (SPC) methodology to ensure that atypical results (even if they are within the wider specification limits) are investigated. However, SPC limits which reflect the achievable process capability (within part of the design space) should not be confused with specification limits.
For API specifications, in addition to having raw material and starting material specifications as the primary control point where possible, intermediate specifications may be simplified. This can lead to reduced testing by concentrating more on the link to CQAs and impurity/solvent purging knowledge across the design space and less on what has been seen in target-point batches.
In summary, control strategy attribute controls for all materials will be scientifically justified by ensuring all of the following aspects are considered and enacted where appropriate:
- Comprehensive raw material and starting material specifications to control CQAs at point of origin
- Intermediate specifications with reduced testing based on link to CQAs and utilizing impurity purging knowledge
- In process monitoring/testing in addition to or as a replacement for end product testing
- API specification limits based on safety and clinical efficacy data, taking into account the limits obtained from manufacturing performance across the defined design space
Similar strategies are also applicable to drug product specifications.
Impact of Specification Changes on Method Capability
Regulatory authorities assess whether the control strategy (including specifications) proposed for the API/drug product and supporting manufacturing processes are adequate.8 For certain methodologies, there are conflicting requirements for allowable limits that can result in specifications being tightened during review. For example, for residual solvents, a gas chromatography method developed with sensitivities aligned with ICH Q3C13 limits will not be fit for purpose if the limits are tightened to reflect typical process values, which are normally significantly lower than the ICH safety-based limits. A similar scenario exists with dissolution methods. There is a general consensus that dissolution testing should be sensitive to relevant changes in the product formulation, be a robust quality control (QC) tool, but also be able to predict in vivo performance of the drug product.15 Dickinson, et al16 indicated that dissolution specification setting was, therefore, often a balancing act between under- and over-discriminating approaches. In the former, there is clear patient risk, while in the latter case there is a clear manufacturer risk with acceptable batches being inappropriately rejected and a significantly reduced process capability for the drug product (as process capability and specifications are intrinsically linked).
Dickinson, et al16 identified possible failure modes and manufactured 3 different drug product variants, based on (i) API particle size differences, (ii) different formulation variants (decreased disintegrant levels and increased binder), (iii) altered processing variants (increased granulating fluid and granulation time). In parallel, 4 dissolution methods were developed based on 3 different aqueous buffers (pH 1.2, 4.5, and 6.8) and a separate surfactant method. The optimal method (demonstrating good discrimination and precision) was the surfactant method. The different manufacturing variants were assessed, together with the standard drug product and an oral solution, in an in vivo relative bioavailability study. Similar pharmacokinetic (PK) behavior was found between the tablet variants, the standard tablet, and the oral solution formulations. Thus, tablets manufactured with the slowest dissolution rate showed similar PK to the standard tablet, and dissolution was, therefore, not a rate-limiting attribute for absorption. Consequently, the design space boundaries for this tablet product could be embraced by the standard tablet and the slowest dissolving tablet variant measured using the surfactant dissolution method.
Dickinson16 indicated that although all dissolution profiles were within the proposed design space and had equivalent bioavailability, the regulatory authorities requested different dissolution methods and different dissolution specification limits for the 2 main global markets (EU and US).
Conclusion
The ATP can be acceptable as a qualifier of the expected method performance by analogy to the QTPP.7 An ATP, however, can only be defined when:
- All the CQAs have been identified.
- A holistic control strategy has been developed which identifies, based on risk, where specification limits and analytical methods are required.
- Specification limits are defined for CQAs based on patient safety and clinical efficacy but also take into account predicted/observed levels of CQAs across the entirety of the design space (rather than a limited number of batches operated at or close to the target parameter setpoints).
During the definition of the control strategy, a generic ATP (specific to what is expected from a particular analytical technique) can be used to support development. This generic ATP will evolve into the commercial ATP once the control strategy is finalized.
There are conflicting requirements for allowable specification limits that can result in limits being tightened during review, and consequently, regulatory authorities requesting different limits (and even different methodologies altogether). This presents a challenge for determining whether analytical methods are fit for purpose as this could lead to the ATP being updated (after method validation has been performed) or even lead to multiple ATPs across different markets. As industry operates globally, there is a need for globally approved specification limits and methodology.
References
- Borman P, Chatfield M, Nethercote P, Thompson D, Truman K. The application of quality by design to analytical methods. Pharmaceutical Technology. 2007;31(10): 142-152.
- Pohl M, Schweitzer M, Hansen G, et al. Implications and opportunities of applying the principles of QbD to analytical measurements. Pharmaceutical Technology Europe. 2010;22(2):29-36.
- Borman P, Chatfield M, Jackson P, Laures A, Okafo G. Reduced method robustness testing of analytical methods driven by a risk-based approach. Pharmaceutical Technology. 2010; 34(4):72-86.
- Borman PJ, Chatfield MJ, Damjanov I, Jackson P. Method ruggedness studies incorporating a risk based approach: A tutorial. Analytica Chimica Acta. 2011;703(2):101-113.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticasl in Human Use. Quality Guideline Q8(R2) Pharmaceutical Development. August 2009.
- Martin GP, Barnett LK, Burgess C, et al. Lifecycle management of analytical procedure: method development, procedure performance qualification, and procedure performance verification.Pharmacopeial Forum. 2013;39(5).
- EMA-FDA pilot program for parallel assessment of Quality-by-Design applications: lessons learnt and Q7A resulting from the first parallel assessment. U.S. Food and Drug Administration, European Medicines Agency. August 20, 2013. Available at: http://www. ema.europa.eu/docs/en_GB/document_library/Other/2013/08/WC500148215.pdf. Accessed January 2, 2015.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Quality Guideline Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). May 2012.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Multidisciplinary Guideline M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potetntial Carcinogenic Risk. June 2014.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Quality Guideline Q3A(R2) Impurities in New Drug Substances. October 2006.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Quality Guideline Q3B(R2) Impurities in New Drug Products. June 2006.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Quality Guideline Q6A Specifications: Test Procedures and acceptance criteria for New Drug Substances and New Drug products: Chemical Substances. October 1999.
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals in Human Use. Quality Guideline Q3C(R5) Impurities: Guideline for Residual Solvents. February 2011.
- Wheeler DJ, Chambers DS. Understanding Statistical Process Control. Vol 37. 2nd ed. Knoxville, TN: SPC press; 1992.
- Anand O. Dissolution testing: An FDA perspective. AAPS Workshop – Physical Pharmacy and Biopharmaceutics, May 13, 2009. Available at: http://mediaserver.aaps.org/meetings/09_ ppb/wed/track_i/om_anand.pdf. Accessed January 2, 2015.
- Dickinson P, Flanagan T, Holt D, Reynold G, Stott P. Therapeutic Equivalence in a Quality by Design World Linking the patient to the quality target product profile. JPAG meeting on Quality by Design, RSC Headquarters, Burlington House, London. 20 March 2014.
Phil Borman is a Chartered Chemist with more than 18 years of experience in the pharmaceutical industry, having obtained a Masters in Chemistry from UMIST (Manchester) University and a Masters in Industrial Data Modelling from De Montfort (Leicester) University. Phil is currently accountable for the provision of QbD Support (with an emphasis on Analytical QbD) across multiple late-phase project teams. Phil pioneered the adaptation of QbD principles to Analytical methods and has published widely in the field of Analytical Chemistry.
Matthew E. Popkin, PhD (University College London), BSc (University of Bristol) is Product Development Regulatory Liaison at GSK based in Stevenage, UK. Matt has worked for GSK since 2000, also being based in that time in Tonbridge, UK and Verona, Italy. In his time at GlaxoSmithKline, he has led project teams responsible for the development and registration of many new API manufacturing processes. Matt’s primary research interest is in the application of Quality by Design to the development and registration of new pharmaceutical products and manufacturing technologies.
Nicola Oxby has a degree in chemistry (BSc) from University of Newcastle upon Tyne and 14 years of experience in the pharmaceutical industry. She is currently a team manager within Product Development in GSK R&D. Other roles within the company have included technology transfer lead for an existing GSK product at a manufacturing site in Ireland and analytical lead for late-phase development products ensuring the QbD aspects of analytical activities were applied.
Marion Chatfield is a statistician with 30 years’ experience working in the pharmaceutical industry, having obtained degrees in Mathematics (MA) and Applied Statistics (MSc). After gaining broad pharmaceutical experience, she has focused her efforts in the last 15 years on the application of statistics in process chemistry and product development. In particular she pioneered the application of QbD principles to analytical methods within GSK and has also published externally.
David Elder has degrees in Chemistry (BSc), analytical chemistry (MSc), and a PhD in Crystallography (Edinburgh). He is a visiting professor at King’s College, London. Dave has 37 years of experience in the pharmaceutical industry. He is currently a director within the Product Development group in GSK R&D. He has 70 publications and has given nearly 100 external presentations at scientific symposia. He has co-edited one book on the Analytical Characterisation and Separation of Oligonucleotides and their Impurities.